Evaluación de la calidad fisicoquÍmica en cápsulas de cefalexina 500 mg
Quality evaluation of cephalexin 500 mg capsules
Samantha Pereira-Leiva1, Esteban Pérez-López2
Fecha de recepción: 20 de junio de 2023
Fecha de aprobación: 6 de octubre de 2023
Pereira-Leiva, S; Pérez-López, E. Evaluación de la calidad fisicoquímica en cápsulas de cefalexina 500 mg . Tecnología en Marcha. Vol. 37, No 2. Abril-Junio, 2024. Pág. 3-14. https://doi.org/10.18845/tm.v37i2.6662
https://doi.org/10.18845/tm.v37i2.6662
Palabras claves
Cefalexina; antibiótico; pruebas de calidad; medicamentos; USP.
Resumen
El presente artículo contiene los detalles sobre el estudio realizado a la calidad fisicoquímica en las cápsulas de cefalexina de 500 mg que se comercializan en Costa Rica. La cefalexina es un antibiótico de uso frecuente que permite combatir las infecciones causadas por bacterias gramnegativas, grampositivas, y es de gran demanda debido a que presenta poca toxicidad y un amplio margen terapéutico según señalan distintos autores. El objetivo de la presente investigación fue determinar la calidad durante su vida de anaquel, de las cápsulas de cefalexina 500 mg, en cuanto a los principales parámetros fisicoquímicos evaluables. La metodología se basó en realizar un muestreo aleatorio a nivel de farmacias de la zona central del país y adquirir la cantidad de dosis (cápsulas) necesarias de un mismo lote de cada una de las marcas que fueran ubicadas en dichos comercios, para proceder a someterlas a las principales pruebas de calidad en el Laboratorio de Química del Recinto de Grecia de la Universidad de Costa Rica según lo establecido en la Farmacopea de los Estados Unidos (USP 43). Los resultados evidencian que, para la prueba de ensayo de principio activo, el contenido de cefalexina en las cinco marcas contempladas está entre 99-103% de lo etiquetado; para la prueba de disolución las cinco marcas oscilaron entre 110-117% de principio activo disuelto de la cantidad declarada en la etiqueta, y en la prueba de uniformidad por variación de peso los resultados reflejan que el valor de aceptación (AV) obtenido oscila entre 3 y 12 en las distintas marcas. En conclusión, se determinó el cumplimiento de las cinco marcas contempladas en cuanto a los criterios USP establecidos: para la prueba de potencia de principio activo 90-120% de lo etiquetado, la prueba de disolución ninguna dosis es menor al 85% (Q+5%) disuelto, y para la prueba de variación de peso el AV ≤ 15.
Keywords
Cefalexin; antibiotic; quality tests; drugs; USP.
Abstract
This article contains details about the study carried out on the physicochemical quality of the 500 mg cephalexin capsules sold in Costa Rica. Cephalexin is a frequently used antibiotic that helps fight infections caused by gram-negative and gram-positive bacteria, and is in great demand because it has little toxicity and a wide therapeutic range, according to different authors. The objective of the present investigation was to determine the quality during its shelf life of cephalexin 500 mg capsules, in terms of the main evaluable physicochemical parameters. The methodology was based on conducting a random sampling at the pharmacy level in the central area of the country and acquiring the necessary number of doses (capsules) from the same batch of each of the brands that were located in said stores, to proceed to submit them. to the main quality tests in the Chemistry Laboratory of the Grecia Campus of the University of Costa Rica laboratory as established in the United States Pharmacopeia (USP 43). The tests were carried out in the Chemistry Laboratory of the Grecia Campus of the University of Costa Rica. The results show that, for the active ingredient test, the cephalexin content in the five brands considered is between 99-103% of labeling; For the dissolution test, the five brands ranged between 110-117% of dissolved active ingredient of the amount declared on the label, and in the uniformity test by weight variation, the results reflect that the acceptance value (AV) obtained ranges between 3 and 12 in the different brands. In conclusion, the compliance of the five contemplated brands was determined in terms of the established USP criteria: for the active ingredient potency test 90-120% of labeling, the dissolution test no dose is less than 85% (Q+ 5%) dissolved, and for the weight variation test the AV ≤ 15.
La cefalexina es un antibiótico utilizado en la medicina para combatir infecciones causadas por microorganismos gram positivos y gram negativos; este tipo de bacterias son agentes patógenos para la salud humana [1], por lo que es importante asegurar buenas prácticas de manufactura, por medio de análisis de control de calidad para garantizar el cumplimiento de las especificaciones establecidas para el fármaco. Cual es la referencia
Este antibiótico pertenece al grupo β-lactámicos que se encuentra dentro del grupo de las cefalosporinas, los cuales se emplean para el tratamiento de infecciones causadas por microorganismos gramnegativos y grampositivos. Específicamente, la cefalexina ayuda a combatir las infecciones causadas por gramnegativos como la Salmonella y Pasteurella, y es de gran demanda debido a que presenta poca toxicidad y un amplio margen terapéutico [1].
Según Gómez, et al. [2] en su estudio sobre los betalactámicos en la práctica clínica, indica que el mecanismo de acción de los β-lactámicos consiste en inhibir la formación de la pared bacteriana, provocando una interferencia en la síntesis del peptidoglicano mediante un bloqueo en la última etapa de su producción (transpeptidación), además actúa activando la autolisina bacteriana endógena que destruye el peptidoglicano, las cuales son bacterias parciales, actuando únicamente con base al crecimiento celular.
Por otra parte, en el plano experimental a nivel de laboratorio, es necesario mencionar que la técnica de espectrofotometría UV-Vis, es una de las más útiles para determinar la calidad en múltiples fármacos, cuando se demuestra que no existe interferencia por co-absorción a una misma longitud de onda de trabajo. Esta técnica analítica consiste en determinar la concentración de analitos en solución, cuyas moléculas absorben radiaciones electromagnéticas y esta energía que absorben en forma de luz está relacionada linealmente con la concentración [3].
En línea con lo anterior, como evidencia de estudios realizados con cefalexina, destaca el trabajo publicado por Chorilli, et al. [4], determinaron el comportamiento del activo cefalexina en diferentes marcas en Brasil, en su investigación titulada “Estudo de perfil de dissolução dos medicamentos de referência, genérico e similar contendo cefalexina na forma farmacêutica cápsula”; cuyo fin consistió en evaluar el perfil de disolución de fármacos de referencia, genéricos y similares que contienen 500 mg de cefalexina en cápsulas. Las muestras fueron sometidas a pruebas de disolución y perfiles de disolución in vitro, concluyendo que las mismas cumplen con las especificaciones y que son de rápida disolución, ya que los porcentajes de disolución fueron mayores al 85%, existiendo una gran similitud entre las curvas obtenidas para cada muestra, sugiriendo que son fármacos equivalentes.
Asimismo, Cuencas [5] en Bolivia, para su tesis de grado de licenciatura en química farmacéutica, sobre el desarrollo y validación de cefalexina comprimidos de 500 mg por espectrofotometría ultravioleta, planteó la investigación-desarrollo-validación de la metodología analítica mediante la espectrofotometría ultravioleta, la metodología empleada consistió primeramente en pruebas al espectrofotómetro como la aptitud de absorbancia, precisión y exactitud fotométrica, posteriormente pruebas de estabilidad a las muestras escogidas así como a los estándares, concluyendo que el método es óptimo, y procedió a la validación del mismo, determinando que el método tiene una buena especificidad al no encontrar diferencias significativas entre las muestras y estándares secundarios; presenta un coeficiente de correlación de 0,998 por ende es lineal, la sensibilidad con relación a exactitud presenta un porcentaje de recuperación dentro del rango establecido, además el método es preciso después de determinarlo con estudios de repetibilidad del instrumento y una presión intermedia bajo un tiempo y espacio diferente.
De forma similar, García et al. [1] en su investigación realizada en Cuba, sobre la evaluación del desempeño del método analítico para la cuantificación de la materia prima de cefalexina, cefaclor, cefoxitina sodica y cefixina, llevó a cabo el estudio utilizando la técnica de cromatografía líquida de alta resolución en fase reversa, la preparación de estándares, muestras y los sistemas de solventes para cada compuesto corresponde a la planteado por la farmacopea oficial, a los datos obtenidos en cada ensayo se le determinó la homogeneidad de varianza para demostrar el comportamiento normal y se aplicó herramientas estadísticas como ANOVA simple, regresión lineal, y para la validación se aplicó los lineamientos establecidos por la Conferencia Internacional de Armonización (ICH). Los resultados que obtuvieron concluyeron que los métodos son lo suficientemente específicos, precisos y exactos, con una respuesta lineal en el rango de concentración de 50 a 150%.
El estudio de fármacos a diferentes marcas comerciales suele hacerse con el fin de encontrar diferencias significativas entre cada una de ellas y corroborar que cumple con los estándares de calidad, tal es el caso de Caseres y Fretes [6] en Paraguay en su investigación sobre la comparación de cefalexina 500 mg nacional e importada, cuyo objetivo de estudio fue comparar los perfiles de disolución de especialidades farmacéuticas nacionales conteniendo cefalexina 500 mg con una importada; la metodología utilizada fue la contenida en la monografía USP 37, realizaron las determinaciones con las técnicas de HPLC y espectrofotometría UV-Vis, y los resultados obtenidos fueron que todos cumplen con los límites establecidos y mostraron un comportamiento de disolución similar.
También, Alarcón y Celaguachay [7] en Ecuador, en su trabajo de campo sobre el estudio comparativo de los parámetros fisicoquímicos de cefalexina 500 mg cápsulas genéricas de dos laboratorios farmacéuticos ecuatorianos frente a medicamento innovador; el método analítico usado corresponde al oficial USP 41, cuantificando el principio activo por HPLC y espectrofotometría UV-Vis, teniendo como resultados para laboratorios G-002, G-003, frente innovador I-001 respectivamente, los siguientes: tiempo de desintegración: 2´ 38”; 2´ 36”; 2´ 39”. Valoración por HPLC: 100,22%; 98,33%, 95,66%. Ensayo de disolución 105,90%; 105,91%; 105,21%. Uniformidad de dosis por variación de peso: L1= 9,55%; L1= 8,93%; L1= 8,93%, concluyendo que los medicamentos genéricos estudiados cumplen los parámetros de control de calidad frente al innovador, según las especificaciones técnicas de la USP correspondiente y presentan mínimas variaciones entre sus análisis.
Por su parte, a nivel de Costa Rica no fue posible ubicar estudios publicados sobre calidad en cápsulas de cefalexina específicamente, pero existen investigaciones publicadas con un enfoque similar al de la investigación evidenciada en este artículo con otro tipo de fármacos. Por ejemplo, Pérez [8] en su estudio sobre el análisis comparativo de la uniformidad de contenido en tabletas de sildefanil (50 mg/tab) de un producto genérico y el medicamento original, determinó que el método empleado fue selectivo, preciso y exacto; y las tabletas ensayadas se analizaron por espectrofotometría ultravioleta visible cuyos resultados obtenidos determinan que las muestras cumplen los límites establecidos en la farmacopea estadounidense para la prueba de uniformidad de contenido.
Pérez y Rojas [9] en su estudio sobre la validación de un método para la cuantificación de acetaminofén en tabletas de 500 mg por espectrofotometría ultravioleta para la prueba de uniformidad de contenido, los parámetros estimados fueron: linealidad, exactitud, repetibilidad y precisión intermedia utilizando como referencia la guía de validación del Ente Costarricense de Acreditación (ECA), para un método normalizado y modificado, y con los resultados obtenidos concluyen que el método es lineal, preciso y exacto.
Por su parte, Castiglioni [10], en su tesis sobre la evaluación del impacto de resultados S2 en las pruebas de disolución de fármacos multiorigen presentes en la lista de medicamentos priorizados y que deben demostrar equivalencia terapéutica in vitro; esta investigación se basó en evaluar si la variabilidad de los resultados de la prueba de disolución de rutina en lotes específicos de medicamentos presentes en la lista de medicamentos priorizados por el Ministerio de Salud tienen impacto en la similitud de los perfiles de disolución comparativos en la toma de decisiones, primeramente selecciono medicamentos disponibles en la CCSS, realizó una validación del sistema y método, concluyendo que ambos son precisos y exactos para desarrollar los perfiles de disolución comparativos con el producto de referencia nacional. De los tres lotes escogidos concluye que en todos los casos se cumple la equivalencia terapéutica in vitro, con respecto a la prueba de disolución correlacionó los valores de Q y confirma que entre mayor es el valor de Q en el lote menor será su factor de diferencia y mayor el de similitud en un perfil de disolución comparativo.
Por otra parte, Pérez et al. [11] en su estudio sobre evaluación de los parámetros de calidad en tabletas de ibuprofeno que se consumen en Costa Rica, las muestras de interés fueron adquiridas de forma aleatoria en diferentes locales comerciales, a diferentes potencias 200, 400 y 600 mg de principio activo con forma farmacéutica de tabletas, el método analítico utilizado como referencia es de la USP 39, del cual incluyeron las pruebas de valoración de principio activo por HPLC, el ensayo de disolución cuantificando por espectrofotometría UV-Vis y la uniformidad de dosis unitaria por variación de peso; los resultados que obtuvieron muestran que los productos sometidos a este estudio son seguros al encontrarse dentro de las especificaciones establecidas a nivel farmacopeico.
Por último, es importante resaltar que, la evaluación de parámetros de desempeño en toda metodología analítica, son importantes porque generan evidencia para demostrar que el método es óptimo para ser aplicado, siempre y cuando cumplan con los criterios de aceptación. En este sentido, las pruebas de verificación al método, como linealidad, repetibilidad y precisión intermedia se basan en considerar el equipo, sistema electrónico, operaciones analíticas y muestras a analizar como un sistema integral [5]. Por tal razón, es esencial garantizar la validez de los resultados de laboratorio a través de procesos rigurosos como los señalados.
Con base en lo anterior, se dimensiona la trascendencia dela investigación particular en el ámbito de la calidad de medicamentos, y además, la proyección existente hacia la sociedad costarricense al contemplar como objetivo la evaluación de la calidad de un fármaco de gran relevancia para la salud de las personas como lo es la cefalexina, realizando la adquisición del medicamento para el estudioen su plena vida de anaquel, lo cual permite una postura investigativa sin intereses particulares y prejuicios y garantiza la objetividad de los resultados.
Materiales y métodos
Se realizó la indagación a nivel de farmacias de la zona central del país para conocer cuáles son las diferentes marcas de cefalexina 500 mg cápsulas que se comercializan en el país, y de esta forma se procedió a la compra autorizada para fines investigativos, por tratarse de un medicamento de venta bajo receta médica, contemplando la adquisición de no menos de 50 cápsulas de cada marca y el mismo lote en cada caso, así como, la adquisición de las cápsulas que distribuye la Caja Costarricense de Seguro Social (CCSS). En el Cuadro 1, se incluye el detalle de las marcas incluidas en el estudio.
Cuadro 1. Información de las marcas comerciales de cefalexina en cápsulas de 500 mg utilizadas para la investigación.
|
Marca/procedencia |
Lote |
|
CCSS |
A07102 |
|
La Santé |
3263621 |
|
ADIUVO |
15202 |
|
Biopharma |
A1093 |
|
MK |
SVF5342 |
Los parámetros evaluados fueron linealidad, por medio de tres curvas de calibración en el rango de trabajo (16 a 26 mg/L); la repetibilidad, evaluando no menos de seis réplicas de cada estándar, uno bajo, uno medio y uno alto del rango de concentraciones definidos para la curva de calibración; y la precisión intermedia, que se realizó bajo las condiciones de repetibilidad pero variando el factor tiempo, cambiando el día de análisis entre la recolección de datos correspondientes para la prueba.
Seguidamente, se procedió a la realización de las pruebas de laboratorio para determinar la calidad de los productos contemplados, para lo cual se tomó como base la metodología de la monografía oficial de la USP 43[12]; para evaluar los parámetros de calidad: ensayo de principio activo: el cual se realiza pesando y cuantificando no menos de tres muestras provenientes del contenido homogéneo de 20 cápsulas del producto; la uniformidad de dosis por variación de peso: que se realiza en primera instancia pesando individualmente el contenido de 10 cápsulas del producto y relacionando estos pesos con respecto al resultado promedio del contenido obtenido de la prueba del ensayo; y por último, la prueba de desempeño disolución con medio: agua 900 mL, aparato 1: canastas rotatorias 100 rpm, tiempo: 30 min; la cual se realiza sometiendo al proceso de disolución seis cápsulas (en primera instancia) del producto en estudio, bajo las condiciones descritas.
El equipo utilizado fue el espectrofotómetro ultravioleta-visible Jasco V-730, a 262 nm, concentración de trabajo 20 mg/L con agua destilada como disolvente, y todas las pruebas fueron ejecutadas en el Laboratorio de Química del Recinto de Grecia, de la Universidad de Costa Rica.
Con respecto a la etapa inicial de evaluación de parámetros de desempeño, las pruebas realizadas demostraron linealidad, precisión y exactitud del método para ser empleado con confianza también para la valoración de principio activo por medio de la técnica de espectrofotometría UV-Vis, la cual está establecida oficialmente en la USP para la cuantificación de cefalexina en la prueba de disolución específicamente.
Por su parte, en relación con la valoración del principio activo en las diferentes marcas comerciales de cefalexina 500 mg/cápsula, en el Cuadro 2, es posible destacar el comportamiento homogéneo de los resultados por cada marca indagada, y los promedios obtenidos oscilando entre 99 y 103% de lo etiquetado entre las marcas analizadas, con desviaciones estándar relativas no mayores a 1,82% como la obtenida en el caso de las cápsulas de la marca ANDIUVO, la precisión de los datos con desviación estándar relativa más baja obtenida para el caso de las cápsulas de Biopharma con 0,21% de desvío relativo. Lo anterior, considerandoque en esta prueba la desviación obedece a la metodología y al trabajo del analista, y no a la variabilidad en el producto, lo cual se evalúa en la prueba de variación de peso.
Cuadro 2. Resultados de la valoración de cefalexina cápsulas en cinco productos comerciales expresado como % de lo etiquetado.
|
Muestra |
CCSS |
La Santé |
ADIUVO |
Biopharma |
MK |
|
1 |
102.71 |
98.35 |
97.07 |
100.95 |
100.23 |
|
2 |
100.78 |
100.10 |
99.82 |
101.35 |
100.88 |
|
3 |
104.11 |
99.87 |
100.46 |
101.24 |
100.80 |
|
Promedio |
102.53 |
99.44 |
99.12 |
101.18 |
100.64 |
|
DSR (%) |
1.63 |
0.95 |
1.82 |
0.21 |
0.35 |
Al evaluar la calidad de los distintos productos comerciales de cefalexina 500 mg/cápsulas, fue posible dimensionar que los resultados reflejan que todas las marcas contempladas en el estudio cumplen con el criterio de aceptación farmacopeico para la prueba de valoración de principio activo, el cual establece que el contenido de cefalexina, en términos de porcentaje de lo etiquetado, debe estar entre 90-120% de potencia del analito [12], lo cual se cumple en todos los casos evaluados con promedios por encima del 99% y por debajo del 103% en todas las marcas (ver Figura 1).
En relación con lo anterior, Rodríguez [13] menciona que el principio activo de un medicamento es la parte esencial de cualquier presentación farmacéutica, la sustancia responsable del efecto farmacológico de un medicamento. Por lo cual, es primordial y obligatorio asegurar que el componente principal del fármaco esté presente en la formulación en el ámbito porcentual que establece la farmacopea oficial, para garantizar el efecto esperado del fármaco al ser suministrado.
Es importante, considerar en la parte de manufactura que los medicamentos siempre oscilen su potencia alrededor del 100% o más de lo etiquetado, y no cercano al límite inferior de la especificación. Si bien para esto existen los estudios de estabilidad, los cuales se basan en evidenciar la capacidad que tiene un medicamento de mantener por un determinado tiempo las características fisicoquímicas originales dentro de las especificaciones [14]; si los fármacos salen a la venta con una potencia cercana al límite inferior de la especificación, podría presentarse que el principio activo llegue a bajar ese porcentaje de lo etiquetado y llegar a no cumplir con su principal función activa dentro de su vida de anaquel.
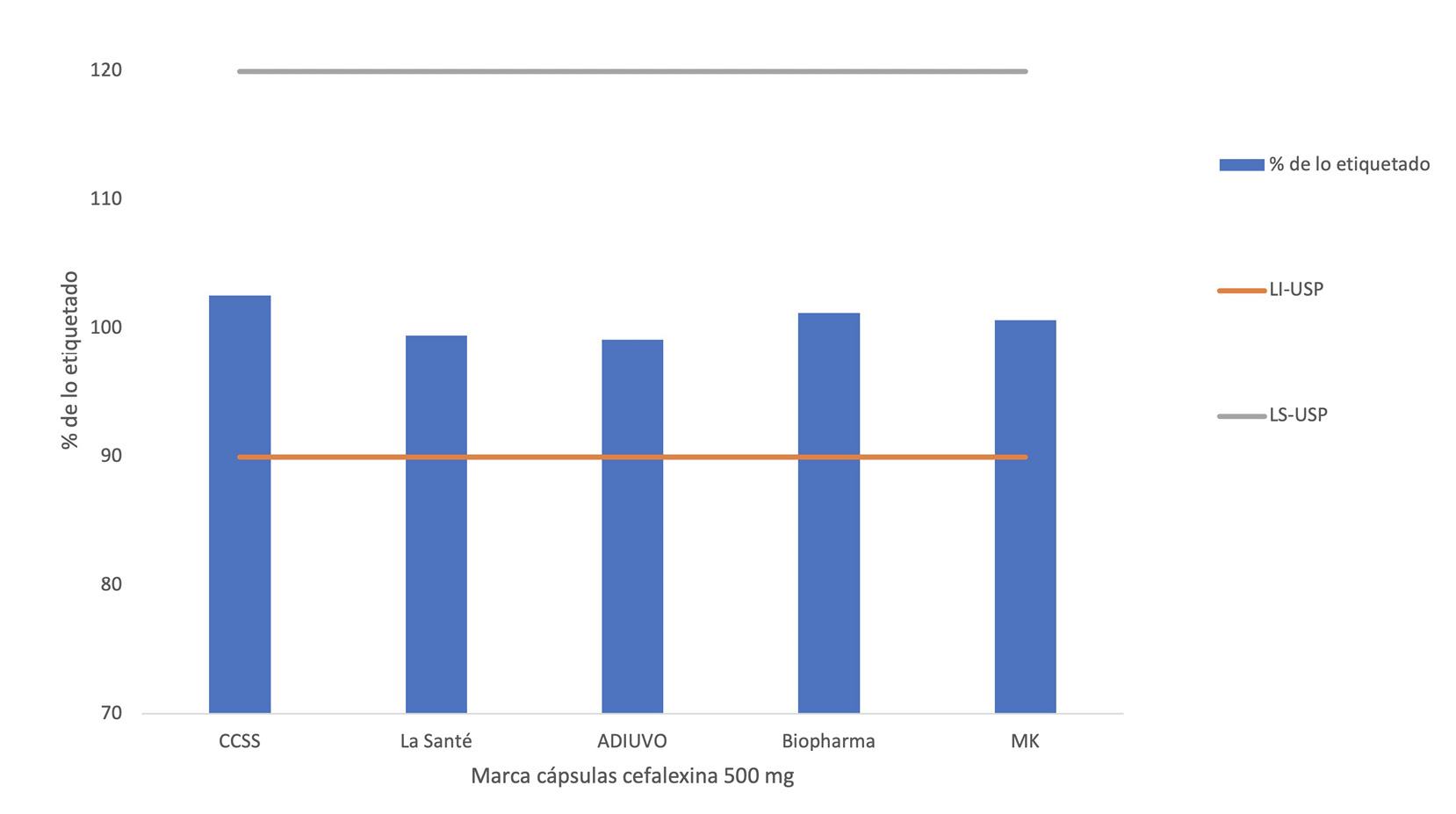
Figura 1. Análisis de resultados para la valoración de principio activo en cefalexina cápsulas 500 mg de distintas marcas
De igual forma, en lo que corresponde a la prueba de disolución; los resultados en el Cuadro 3, permiten destacar que todas las cápsulas individuales, sin importar la marca, se disolvieron por encima del 100% con respecto a la cantidad declarada en la etiqueta (500 mg); y los promedios de porcentaje disuelto por marca oscilan entre 110% y 117%, con desviaciones estándar relativas entre 1,07% en el caso de la marca La Santé y 4,90% en las cápsulas que distribuye la CCSS.
Cuadro 3. Resultados de la prueba de disolución para las cinco marcas comerciales de cefalexina cápsulas expresado como % disuelto.
|
CCSS |
La Santé |
ADIUVO |
Biopharma |
MK |
|
|
1 |
100.01 |
117.27 |
104.44 |
113.51 |
113.73 |
|
2 |
109.31 |
117.49 |
111.52 |
116.83 |
118.82 |
|
3 |
113.29 |
115.95 |
112.85 |
115.28 |
119.26 |
|
4 |
114.18 |
117.05 |
112.41 |
115.50 |
107.98 |
|
5 |
109.75 |
115.50 |
107.98 |
117.94 |
118.38 |
|
6 |
114.18 |
119.04 |
111.08 |
113.95 |
112.41 |
|
Promedio |
110.12 |
117.05 |
110.04 |
115.50 |
115.64 |
|
DSR (%) |
4.90 |
1.07 |
2.94 |
1.45 |
3.90 |
Estos resultados obtenidos para la prueba de disolución; en las cápsulas de cefalexina de las cinco marcas analizadas; hacen posible visualizar que los resultados son satisfactorios en vista del cumplimiento con respecto al criterio de aceptación definido por la farmacopea, el cual establece que la disolución del fármaco no puede ser menor a Q+5%, (donde Q=80%) para cada unidosis ensayada, en un tiempo de 30 minutos definidos para la prueba según la monografía específica [12] y como se aprecia en los resultados, todas las marcas tienen porcentajes de disolución por encima del 100% disuelto con respecto a lo etiquetado (ver Figura 2); por lo que, su uso es confiable para las personas en términos de asegurar la solubilidad del fármaco en el organismo y cumplir el efecto terapéutico para el cual se suministra.
Con respeto a la prueba de disolución, la Dirección de Regulación de la Salud [15], hace referencia a que la prueba de rendimiento sirve para la determinación de la velocidad de disolución de un medicamento empleando los criterios específicos definidos para cada medicamento en específico.
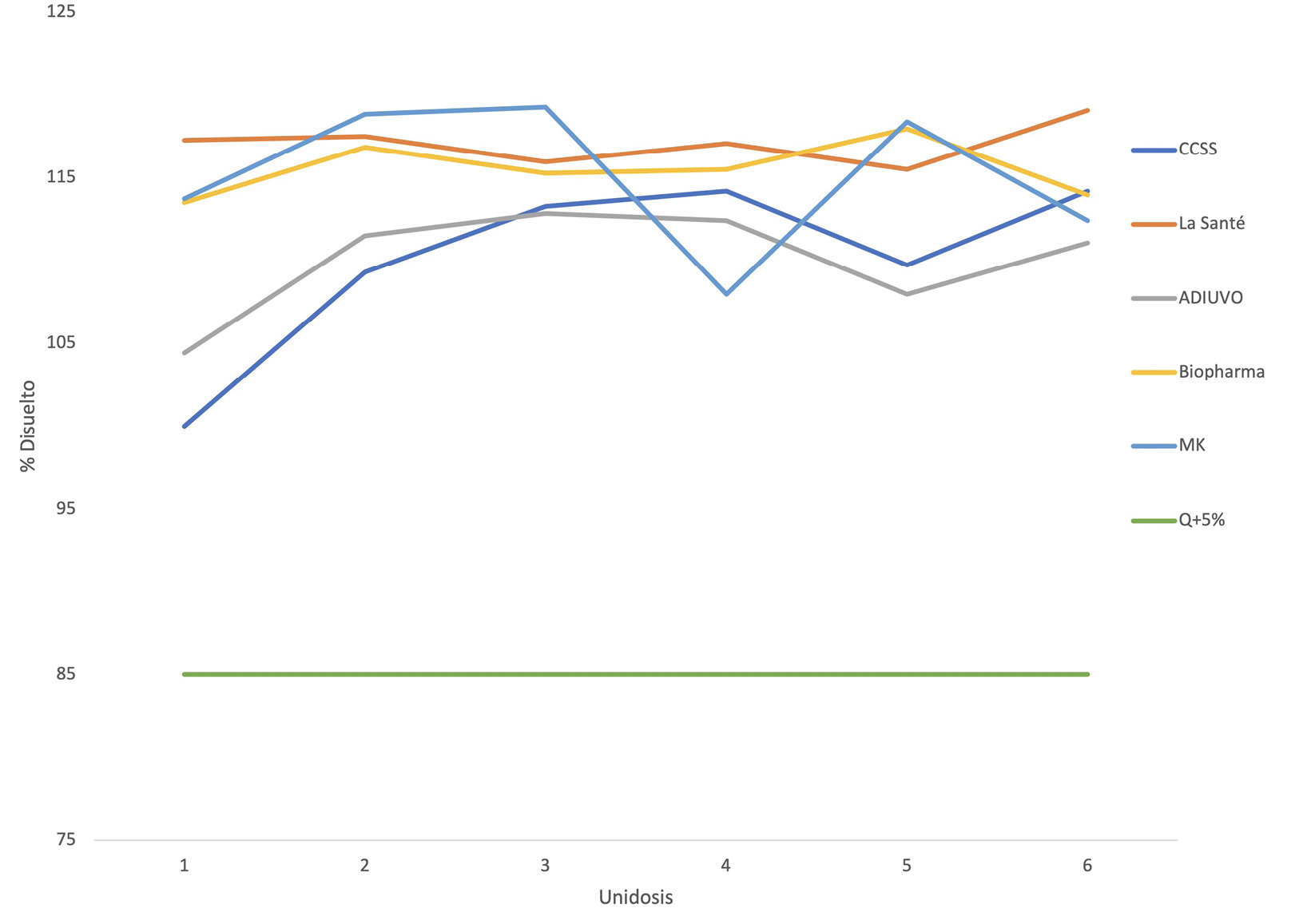
Figura 2. Análisis de resultados para la prueba de disolución de cefalexina cápsulas 500 mg de distintas marcas.
Por último, en lo que respecta a los resultados de la uniformidad de dosis unitaria por variación de peso, en el Cuadro 4, se puede destacar que los pesos obtenidos para las cápsulas individuales, sin importar marca, representan contenido de principio activo no menor a 93,77% de lo etiquetado como en el caso de una cápsula de la marca Biopharma y alcanza valores de hasta 108,86% de lo etiquetado como en el caso de una de las cápsulas de la marca que distribuye la CCSS. Tales diferencias se deben principalmente a variantes particulares del proceso de manufactura de cada lote de cápsulas de cefalexina según la marca del producto.
Por su parte, en cuánto a la variabilidad presentada, lo cual representa la esencia que pretende medir esta prueba, se obtuvieron desviaciones entre 1,39% en el caso de la marca La Santé y 4,55% para las cápsulas de la marca ADIUVO y con respecto al valor de aceptación AV este osciló entre 3,3 y 11,19 entre las distintas marcas, siendo el más bajo el obtenido para la marca La Santé y el AV más alto el de la marca que distribuye la CCSS.
Cuadro 4. Uniformidad de dosis por variación de peso en cefalexina cápsulas expresado como % de lo etiquetado.
|
Muestra |
CCSS |
La Santé |
ADIUVO |
Biopharma |
MK |
|
1 |
105.26 |
96.96 |
98.60 |
93.77 |
101.01 |
|
2 |
105.66 |
99.70 |
100.64 |
100.97 |
98.50 |
|
3 |
96.85 |
100.57 |
102.72 |
101.94 |
103.51 |
|
4 |
99.31 |
99.72 |
101.27 |
101.38 |
100.11 |
|
5 |
97.23 |
97.07 |
94.04 |
100.80 |
100.43 |
|
6 |
106.37 |
97.42 |
101.63 |
102.49 |
103.16 |
|
7 |
104.93 |
99.37 |
107.74 |
103.40 |
99.13 |
|
8 |
102.38 |
100.55 |
95.01 |
102.20 |
99.09 |
|
9 |
108.86 |
99.79 |
94.34 |
102.77 |
103.98 |
|
10 |
103.14 |
98.85 |
95.01 |
100.27 |
101.08 |
|
Promedio |
103.00 |
99.00 |
99.10 |
101.00 |
101.00 |
|
DSR% |
3.92 |
1.39 |
4.55 |
2.69 |
1.94 |
|
AV |
11.19 |
3.3 |
10.8 |
6.52 |
4.97 |
Esta prueba se lleva a cabo para determinar la variabilidad en el contenido de principio activo por unidad de dosificación en un lote dado, y cada unidad ensayada en el lote específico debe tener un contenido de fármaco dentro de un intervalo estrecho alrededor de la cantidad declarada. En este caso, todas las cápsulas de las distintas marcas consideradas cumplen con el criterio para el valor de aceptación definido para la prueba según la farmacopea oficial, el cual se establece en un AV no mayor a 15 [12] para 10 cápsulas analizadas en cada producto de las distintas marcas (ver Figura 3).
Considerando lo anterior, el cumplimiento de la prueba de uniformidad brinda garantía al consumidor, de que, al proceder a la ingesta del fármaco medicado, cada cápsula contiene al menos,lo mínimo necesario de principio activo para recibir el efecto curativo deseado al ingerir según la indicación médica, y que, no contiene más de lo recomendado de principio activo para no ocasionar afectación por sobredosis y los efectos indeseados que esto pudiera generar.
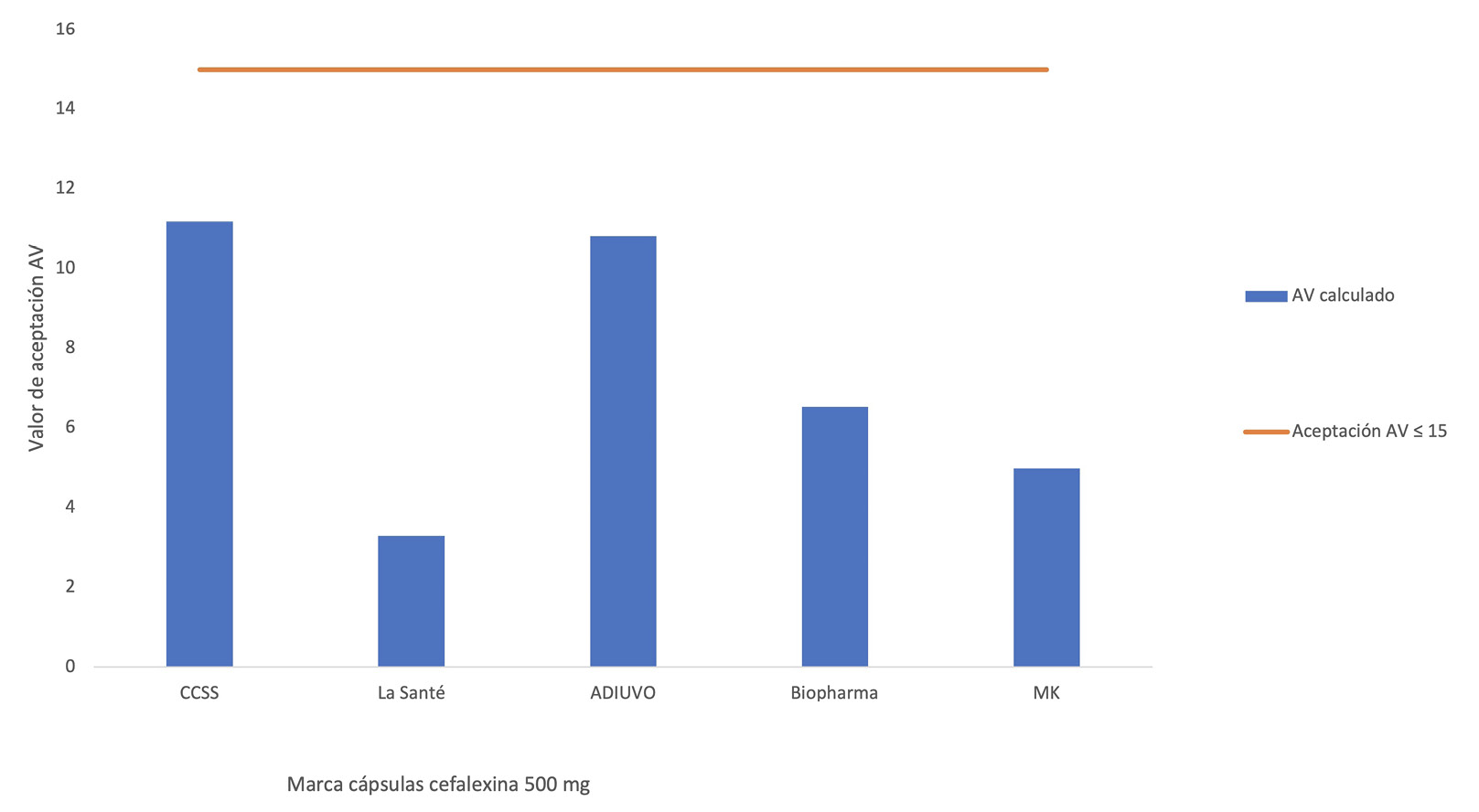
Figura 3. Análisis de resultados para la prueba de uniformidad por variación de peso en cefalexina cápsulas 500 mg de distintas marcas
No existe una regla que regule el peso que deben poseer las dosis de un medicamento determinado, ya que queda a disposición de cada laboratorio fabricante estipular el peso de la unidosis, el cual puede variar debido a diversos factores como: tipo, cantidad y voluminosidad de los excipientes utilizados en la formulación; y el estricto cumplimiento de las buenas prácticas de fabricación durante el proceso de manufactura aseguran la uniformidad del peso en la tableta [16]. Lo que si está claro, es que la cantidad de principio activo, más allá del peso total de la unidosis, debe estar contemplado dentro de lo establecido a nivel de las farmacopeas oficiales, y al ser suministrado el fármaco, este nunca debe sobrepasar la recomendación médica para lograr el efecto terapéutico previsto sin afectación colateral más allá de lo esperado.
•Considerando la prueba de calidad farmacopeica de valoración de principio activo ensayada a las cápsulas de cefalexina 500 mg, realizada a muestras de un lote específico de cada uno de los cinco productos contemplados en la investigación, se establece que todas las marcas cumplen con los criterios de aceptación definidos oficialmente para el contenido de principio activo en cada producto: 90-120% de lo etiquetado, lo cual asegura la cantidad de principio activo necesario para ejercer el efecto farmacológico indicado.
•En lo que respecta a la prueba de disolución realizada según los estándares y parámetros oficiales USP, a seis unidosis de cada marca contemplada, se establece el cumplimiento del criterio de aceptación al obtener que cada cápsula sometida al proceso de disolución logró alcanzar la disolución de la cefalexina en no menos de Q + 5%, donde Q = 80%.
•La prueba oficial farmacopeica de uniformidad de unidades de dosificación por variación de peso, a diez cápsulas de cada una de las cinco marcas contempladas en el estudio, fue posible establecer el cumplimiento de los criterios oficiales establecidos en cuanto a que la estimación del valor de aceptación AV no fue mayor a 15 en todos los productos contemplados, lo cual se relaciona directamente con poca variabilidad en el contenido de cefalexina entre las diferentes unidosis de un mismo producto en un mismo lote.
Agradecimientos
A la Universidad de Costa Rica, Sede de Occidente, Recinto de Grecia.
Participación de autores
EPL: desarrolló el diseño metodológico; SPL: realizó la recolección y análisis de datos;
EPL y SPL: redactaron el manuscrito.
Conflicto de interés
Los autores declaran no tener conflicto de interés.
[1] García Borges, L., García Peña, C., López Armas, M., Martínez Espinosa, V., Fernández Martínez, A., & Cárdenas Peña, M. Evaluación del desempeño del método analítico para la cuantificación de la materia prima de cefalexina, cefaclor, cefoxitina sódica y cefixima. Revista Cubana de Farmacia. 2016; 50(3). Obtenido de: http://www.revfarmacia.sld.cu/index.php/far/article/view/36/40
[2] Gómez, J.; García, E. & Hernández, A. Los betalactámicos en la práctica clínica. Revista Esp Quimioter. 2015; 28 (1):1-9. Obtenido de: https://seq.es/wp-content/uploads/2015/02/seq_0214-3429_28_1_gomez.pdf
[3] Gómez, M., & Tito, V. Análisis multivariado de componentes terpénicos, en aceite esencial de hierba luisa (Cymbopogon citratus), mediante espectrofotometría UV-Visible Derivada (Bachelor’s thesis). 2017. Obtenido de: https://dspace.ups.edu.ec/handle/123456789/14765
[4] Chorilli, M; Souza, A.; Corrêa, F & Salgado, H. Estudo de perfil de dissolução dos medicamentos de referência, genérico e similar contendo cefalexina na forma farmacêutica capsula. Revista de ciências farmacêutica básica e aplicada. 2010; 31(1):69-73. Obtenido de: http://rcfba.fcfar.unesp.br/index.php/ojs/article/view/413
[5] Cuencas, S. Desarrollo y Validación de un método analítico para la valoración de cefalexina comprimidos de 500 mg por espectrofotometría ultravioleta. Universidad Mayor de San Andrés. Bolivia. 2016. Obtenido de: https://repositorio.umsa.bo/bitstream/handle/123456789/17355/T-1900.pdf?sequence=2&isAllowed=y
[6] Caseres, M. & Fretes, S. Comparación de la cefalexina 500 mg nacional e importada en Paraguay. Revista de Ciências de la salud UDES. 2017; 4(2):85-89. Disponible DOI:10.20320/rfcsudes.v4i2.204Alarcón, A. & Celaguachay, C. Estudio comparativo de los parámetros fisicoquímicos de cefalexina 500 mg cápsulas genéricas de dos laboratorios farmacéuticos ecuatorianos frente al medicamento innovador. Universidad de Guayaquil. Ecuador. 2021; Obtenido de: http://repositorio.ug.edu.ec/handle/redug/50868
[7] Pérez, E. Análisis comparativo de la uniformidad de contenido en tabletas de sildenafil (50 mg/tab) de un producto genérico y el medicamento original. Rev. Pensamiento Actual. Universidad de Costa Rica. 2015; Vol. 14 N.23. pp.71-80. Obtenido de: https://revistas.ucr.ac.cr/index.php/pensamientoactual/article/view/19195/19281
[8] Pérez, E. & Rojas, A. Validación de un método para la cuantificación de acetaminofén en tabletas de 500 mg por espectrofotometría ultravioleta para la prueba de uniformidad de contenido. Revista Intersedes. 2016. Disponible: https://doi.org/10.15517/isucr.v17i35.25563
[9] Pérez, E.; Cordero, J. & Bastos, H. Evaluación de los parámetros de calidad en tabletas de ibuprofeno que se consumen en Costa Rica. Revista Pensamiento actual. Universidad de Costa Rica. 2020; 20 (34). 99-114. Disponible DOI: 10.15517/PA.V20I34.41787USP 43 - NF 38. Farmacopea de los Estados Unidos de América. Rockville, Maryland: The United States Pharmacopeial Convention. 2019.
[10] Rodríguez, A. El valor del principio activo. Engenerico. 2016. Obtenido de: https://www.engenerico.com/el-valor-del-principio-activo/
[11] Mendoza, O. Estabilidad de medicamentos. Bmeditores. 2021. Obtenido de: https://bmeditores.mx/entorno-pecuario/estabilidad-de-medicamentos/
[12] Dirección de Regulación de la Salud. Guía para la verificación de la calidad de los medicamentos. 2009. Obtenido de: https://admin.colfar.com/index.php?option=com_phocadownload&view=category&download=409:productos-farmaceuticos-medicamentos-para-uso-humano-verificacion-de-la-calidad&id=9:leyes-y-reglamentos&Itemid=217&start=20
[13] Awofisayo, S., Willie, E. & Umoh, E. Quality Control Evaluation of Multi-Source Artemether-Lumefantrine Tablets Prescribed for Uncomplicated Multi-drug Resistant Malaria. Indian Journal of Novel Drug Delivery. 2010; 2(4):153-157. Obtenido de: https://www.cabdirect.org/cabdirect/abstract/20113162452
[14] Mendoza, O. Estabilidad de medicamentos. Bmeditores. 2021. Obtenido de: https://bmeditores.mx/entorno-pecuario/estabilidad-de-medicamentos/
[15] Dirección de Regulación de la Salud. Guía para la verificación de la calidad de los medicamentos. 2009. Obtenido de: https://admin.colfar.com/index.php?option=com_phocadownload&view=category&download=409:productos-farmaceuticos-medicamentos-para-uso-humano-verificacion-de-la-calidad&id=9:leyes-y-reglamentos&Itemid=217&start=20
[16] Awofisayo, S., Willie, E. & Umoh, E. Quality Control Evaluation of Multi-Source Artemether-Lumefantrine Tablets Prescribed for Uncomplicated Multi-drug Resistant Malaria. Indian Journal of Novel Drug Delivery. 2010; 2(4):153-157. Obtenido de: https://www.cabdirect.org/cabdirect/abstract/20113162452
1 Universidad de Costa Rica, Sede de Occidente - Recinto Grecia. Correo electrónico: samantha.pereira@ucr.ac.cr
 https://orcid.org/0000-0003-3739-1036
https://orcid.org/0000-0003-3739-1036
2 Universidad de Costa Rica, Sede de Occidente - Recinto Grecia. Correo electrónico: esteban.perezlopez@ucr.ac.cr
https://orcid.org/0000-0002-6379-7139