Identificación de explosivos Orgánicos en Indicios Post-Explosión mediante gc-ms y gc-npd
Identification of Organic Explosives Clues Post-Explosion by Gc-Ms And Gc-Npd Methods
Stephanie Arce-Rubí1, Steven Vargas-Ramírez2, Efraín Solís-Montiel3, Gilberto Piedra-Marín4, Jose Carlos Mora-Barrantes5,,
José Pablo Sibaja-Brenes6, Henry Borbón-Alpizar7
Arce-Rubí, S; Vargas-Ramírez, S; Solís-Montiel, E; Piedra-Marín, G; Mora-Barrantes, J.P; Sibaja-Brenes, J; Borbón-Alpizar, H. Identificación de explosivos orgánicos en indicios Post-Explosión mediante gc-ms y gc-npd. Tecnología en Marcha. Vol. 33-3. Julio-Setiembre 2020. Pág 25-44. https://doi.org/10.18845/tm.v33i3.4377
https://doi.org/10.18845/tm.v33i3.4377
Fecha de recepción: 7 de julio de 2019
Fecha de aprobación: 14 de octubre de 2019
Palabras clave
Explosivos; detonaciones; cromatografía; componentes explosivos; validación.
Resumen
Se implementó y validó un método para la identificación de explosivos por medio de cromatografía de gases acoplada a un detector de espectrometría de masa (GC-MS) y por cromatografía de gases de doble columna acoplada a dos detectores de fósforo-nitrógeno (GC-NPD). El método establecido incluyó el análisis de sustancias explosivas (analitos )de diferente naturaleza química)como TNT, TNB, 2,4-DNT, 2,6-DNT, 2-NT, 3-NT, 4-NT, PETN, NG, RDX y tetril, entre otros. Se evaluó para cada analito el límite de detección, la variabilidad en el tiempo de retención en términos de repetibilidad y precisión intermedia, en ambas técnicas cromatográficas. Utilizando GC-MS las correlaciones encontradas entre los espectros de masas de los patrones y los de los analitos fueron superiores al 90 % para la mayoría de los nitrotoluenos y los estabilizantes de pólvora libre de humos. El análisis por GC-NPD se presentó como método alterno para la identificación de los mismos analitos que se determinan por GC-MS, exceptuando el PETN y el 1,3,5-TNB. Los límites de detección fueron inferiores en uno o dos órdenes de magnitud en relación con los alcanzados por GC-MS y la precisión de los tiempos de retención resultó ser satisfactoria para un análisis cromatográfico. Finalmente, se probó la veracidad y la efectividad de los métodos mediante el análisis de indicios post-explosión obtenidos de las explosiones controladas de seis artefactos explosivos improvisados con cargas explosivas de composición conocida. Los procesos de extracción de las muestras mediante solventes, seguidos por el análisis preliminar de las muestras por TLC y el análisis por GC-MS y GC-NPD permitieron realizar la identificación de al menos un componente explosivo en el 83 % de los casos.
Keywords
Explosives; detonations; chromatography; explosive components; validation.
Abstract
Methods for the identification of explosives by means of Gas Chromatography Mass Spectrometry (GC-MS) and Gas Chromatography with Nitrogen Phosphorus Detectors (GC-NPD) were implemented and validated. The methods established included the analysis of the following explosive substances: TNT, TNB, 2,4-DNT, 2,6-DNT, 2-NT, 3-NT, 4-NT, PETN, NG, RDX and tetril. For each analyte, the detection limit, the variability in retention time for each analyte, in terms of repeatability and intermediate precision were evaluated by using both chromatography techniques. By means of GC-MS the correlations found between the mass spectrum of the standards and those of the analytes went over 90% for the majority of the nitrotoluenes and for some stabilizers of smoke-free gunpowder. The GC-NPD analysis is an alternative method for the identification of the same analytes done by using GC-MS, with the only exceptions of PETN and 1,3,5 -TBN. The detection limits of these two compounds were inferior with respect to the ones reached by GC-MS. Finally, veracity and effectiveness for both methods were proven by analysing post-explosion samples of six explosive materials. All the explosion events were developed under controlled situations. The samples were extracted through organic solvents; then they were analysed by TLC, GC-MS and GC-NDP. This methods allowed the identification at least of one explosive component in the majority of the samples (83%).
Introducción
Según Akhavan [1] y Meyer et ál [2], los explosivos son sustancias químicas puras o mezcladas que por la acción de un estímulo externo experimentan un cambio químico sin una fuente externa de oxígeno, con liberación de grandes cantidades de energía y evolución de gases calientes, todo en un tiempo muy breve. Meyer et ál [2] y Beveridge [3] señalan que la capacidad de un explosivo para descomponerse obedece a reordenamientos internos favorecidos termodinámica- y entrópicamente, considerando la Akhavan [1] divide el proceso de explosión en tres tipos: explosiones físicas, explosiones atómicas y explosiones químicas.
En relación a las explosiones químicas, Venugopalan [4] considera explosiva una sustancia química que tiene una alta energía potencial y puede liberarla en un período muy corto de tiempo. Venugopalan [4] y Urbanski [5] atribuyen las propiedades explosivas químicas a un fenómeno molecular. Warey [6] agrupa los explosóforos en categorías como Nitro (Ej.: 2,4,6 trinitrotulueno, TNT), Nitroester (Ej.: nitroglicerina), Clorato (Ej.: clorato de potasio), Perclorato (Ej.: perclorato de amonio), Acetiluro (acetiluro de cobre), Peróxido (triperóxido de triacetona) y Azida (fulminato de mercurio).
Según la Encyclopedia of Analytical Chemistry [7] la importancia del análisis de los explosivos radica principalmente en su uso legal, por ejemplo, en la industria o el ámbito militar, según la coyuntura gubernamental de cada país. El uso de explosivos es ilegal en el terrorismo y en la actividad criminal. Peterson [8] indica que en los camposmilitar e industrial los explosivos se analizan de forma rutinaria después de su producción, para el control de calidad, siendo relevante no solo el análisis cualitativo, sino también la cuantificación de las sustancias explosivas e impurezas.
Con respecto al impacto ambiental de los explosivos, Hewitt [9] señala que la Agencia de Protección Ambiental de los Estados Unidos, US Environmental Protection Agency (EPA, por sus siglas en inglés), ha establecido algunos métodos para el análisis de explosivos en suelos y en aguas a través de técnicas cromatográficas y de inmunoensayos, como las siguientes [10-17]: 8515/EnSys RIS (método de detección colorimétrico), 8510/EnSys RIS (método de detección colorimétrico), 4050/D TECH (inmunoensayo), 4051/D TECH (inmunoensayo), 8330A y 8330B (HPLC), 8095 (cromatografía de gases) y 529 (cromatografía de gases y espectrometría de masas). Entre algunos analitos que se puede analizar con las técnicas anteriores están los siguientes: TNT 2,4,6 trinitrotolueno, TNB: 1,3,5-trinitrobenceno, DNB: dinitrobenceno, DNT: dinitrotolueno, RDX: ciclotetrametilentrinitramina, HMX: ciclotetrametilentetranitramin, PETN: tetranitrato de pentaeritrita¸ NQ: nitroguanidina, NC: nitrocelulosa, NG: nitroglicerina y NT= nitrotolueno.
Schade [18] y Varga [19] indican que existen diferentes técnicas utilizadas para el análisis de explosivos, algunas de las cuales se usan para el análisis in situ y detección de explosivos en tiempo real. Respecto a las técnicas analíticas Byall [20] Royds [21], Garrido [22] y Rhykerd [23] señalan las siguientes: cromatografía de gases acoplada a un analizador térmico de energía (GC-TEA), cromatografía líquida de alta eficiencia, ultravioleta, acoplada a un detector con arreglo de diodos (HPLC-UV o DAD), cromatografía de gases acoplada a detector selectivo de masas (GC-MS), y cromatografía líquida acoplada a un detector selectivo de masas (LC-MS). Entre los tipos de explosivos que se puede analizar con estas técnicas los autores mencionados indican algunos grupos: halógenos, peróxidos, quinonas y compuestos del grupo nitro, orgánicos, plastificantes y estabilizantes, e inorgánicos.
El proyecto de investigación que aquí se presenta tuvo como objetivo la identificación química de los explosivos de tipo orgánico más comúnmente utilizados en las composiciones explosivas (principalmente en los explosivos propelentes y los explosivos militares) utilizando las técnicas de análisis de cromatografía de gases acoplada a detector selectivo de masas (GC-MS) y cromatografía de gases acoplada a detector de fósforo-nitrógeno (GC-NPD), con el fin de verificar cuál es más eficiente en función de los diferentes parámetros de desempeño evaluados.
Metodología
Equipos analíticos
Cromatógrafo de gases con detector selectivo de masas (GC-MSS)
Se utilizó un cromatógrafo de gases, marca Agilent Technologies, modelo 7890A, el cual cuenta con un automuestreador y autoinyector marca Agilent Technologies, modelo 7693, acoplado a un detector de espectrometría de masas inerte con un cuadrupolo, marca Agilent Technologies, modelo 5975C. Se utilizó una columna HP-5MS (de bajo sangrado, 5 % de fenilpolisiloxano y 95 % metilpolisiloxano) de 30 m de longitud, 0,25 mm de diámetro interno y una película enlazada de 0,25 μm de espesor, marca Agilent Technologies J&W. En el puerto de inyección, el equipo posee un liner tipo Split, de 4 mm de diámetro interno, con lana de vidrio desactivada y septa Advance Green. El gas de arrastre utilizado fue helio, de ultra alta pureza (UAP), proveniente de un cilindro marca Praxair de The Balbach Company.
Cromatógrafo de gases con doble detector de nitrógeno-fósforo (GC-NPD)
Se utilizó un cromatógrafo de gases, marca Agilent Technologies 6890+Plus; el equipo cuenta con un automuestreador y autoinyector marca Agilent Technologies, modelo 7693. El cromatógrafo cuenta con doble columna; la primera de ellas es una columna capilar HP- 5MS de 30 m de longitud con una película enlazada de 0,25 μm y diámetro interno de 0,25 mm, marca Agilent Technologies, acoplada al detector NDP2 A, y la segunda es una columna capilar J&W 122-0732 [(14 % cianopropil-fenil)-metilpolisiloxano (DB-1701)] de 30 m de longitud con una película enlazada de 0,25 μm x 0,25 mm de diámetro, acoplada al detector NPD1 B; cada detector de nitrógeno y fósforo (NPD) es de la marca Agilent Technologies.
Los gases utilizados fueron hidrógeno y helio, de ultra alta pureza (UAP), provenientes de un cilindro marca Praxair de The Balbach Company; nitrógeno proveniente de un generador marca Dominick Hunter, modelo G2-110W, y aire procedente de un generador marca Dominick Hunter, modelo UHP-35ZA-S. El puerto de inyección posee un liner splitless de 4 mm de diámetro interno, con lana de vidrio desactivada y septa Advance Green.
Reactivos químicos
Los siguientes son los reactivos y productos químicos utilizados: acetato de etilo (grado pesticida, Fisher Scientific), acetona (grado HPLC, Merck), acetonitrilo (grado HPLC, Merck), etanol (calidad A.C.S, J.T. Baker), hidróxido de potasio (calidad A.C.S, J.T. Baker), reactivo de Griess, tricloroetano (Merck) y una mezcla de hidróxido de potasio en etanol al 5 % en volumen.
Materiales de referencia
Los siguientes son los materiales de referencia utilizados: 1,3,5-trinitrobenceno 1000 μg/mL en acetonitrilo ( SUPELCO), 2,4,6-trinitrotolueno 1000 μg/mL en acetonitrilo (SUPELCO), 2,4-dinitrotolueno 1000 μg/mL en acetonitril (SUPELCO), 2,6-dinitrotolueno 1000 μg/mL en acetonitrilo (SUPELCO ), 2-nitrotolueno 1000 μg/mL en metanol:acetonitrilo (1:1) (AccuStandard ), 3-nitrotolueno 1000 μg/mL en metanol:acetonitrilo (1:1) (AccuStandard ), 4-nitrotolueno 1000 μg/mL en metanol:acetonitrilo (1:1) (AccuStandard), HMX 1000 μg/mL en metanol:acetonitrilo (1:1) (AccuStandard ), nitroglicerina al 1 % en masa (Merck), PETN 1000 μg/mL en acetonitrilo (Cerilliant), RDX 1000 μg/mL en metanol:acetonitrilo (1:1) (AccuStandard) y tetril 1000 μg/mL en acetonitrilo (SUPELCO.).
Curva de calibración
Se prepararon disoluciones madre a una concentración de 200 mg/L de cada analito, utilizando acetonitrilo grado HPLC como disolvente. Además, se prepararon seis disoluciones a partir de las disoluciones madre de 200 mg/L de cada analito para ser utilizadas en el GC-MS, según criterios tomados al inyectar individualmente cada analito y conociendo los tiempos de retención.
Las mezclas se establecieron de la siguiente forma:
- Mezcla GC-MS-1: 2,4-dinitrotolueno, 2,6-dinitrotolueno, nitroglicerina, etilcentralita, difenilamina, N-NO-difenilamina, 4-amino-2,6-dinitrotolueno y 2-amino-4,6-dinitrotolueno, en acetonitrilo. Se prepararon calibradores a las siguientes concentraciones: 4 mg/L, 6 mg/L, 8 mg/L, 10 mg/L y 15 mg/L.
- Mezcla GC-MS-2: TNT, 2-NT, 3-NT, 4-NT, 4-amino-2,6-dinitrotolueno y 2-amino-4,6-dinitrotolueno, en acetonitrilo. Se prepararon calibradores a las siguientes concentraciones: 4 mg/L, 6 mg/L, 8 mg/L, 10 mg/L y 15 mg/L.
- Mezcla GC-MS-3: RDX y 4-amino-2,6-dinitrotolueno, en acetonitrilo. Se prepararon calibradores a las siguientes concentraciones: 75 mg/L, 100 mg/L, 125 mg/L y 150 mg/L.
- Mezcla GC-MS-4: TNB y 4-amino-2,6-dinitrotolueno, en acetonitrilo. Se prepararon calibradores a las siguientes concentraciones: 75 mg/L, 100 mg/L, 125 mg/L y 150 mg/L.
- Mezcla GC-MS-5: PETN y 4-amino-2,6-dinitrotolueno, en acetonitrilo. Se prepararon calibradores a las siguientes concentraciones: 75 mg/L, 100 mg/L, 125 mg/L y 150 mg/L. Mezcla GC-MS-6: Tetril y 4-amino-2,6-dinitrotolueno, en acetonitrilo. Se prepararon calibradores a las siguientes concentraciones: 75 mg/L, 100 mg/L, 125 mg/L y 150 mg/L.
También, se prepararon dos mezclas a partir de las disoluciones madre de 200 mg/L de cada analito para ser utilizadas en el GC-NPD, separadas de la siguiente forma:
- Mezcla GC-NPD-1: 2,4-dinitrotolueno, 2,6-dinitrotolueno, nitroglicerina, etilcentralita, difenilamina, N-NO-difenilamina, 4-amino-2,6-dinitrotolueno y 2-amino-4,6-dinitrotolueno, en acetato de etilo. Se prepararon calibradores a las siguientes concentraciones: 4 mg/L, 6 mg/L, 8 mg/L, 10 mg/L y 15 mg/L a partir de las disoluciones madre preparadas en acetonitrilo. Fueron preparadas con un contenido de un 10% en volumen de nitrógeno como máximo, para que no interfiriera en el análisis.
- Mezcla GC-NPD-2: TNT, 2-NT, 3-NT, 4-NT, Tetril, RDX, 4-amino-2,6-dinitrotolueno y 2-amino-4,6-dinitrotolueno, en acetato de etilo. Se prepararon calibradores a las siguientes concentraciones: 4 mg/L, 6 mg/L, 8 mg/L, 10 mg/L y 15 mg/L, considerando el mismo porcentaje máximo en volumen de nitrógeno que la mezcla GC-NPD-1.
Separación cromatográfica
Para el análisis de los materiales explosivos mediante el GC-MS, se tomó como referencia las condiciones cromatográficas de operación descritas por la US Enviromental Protection Agency [17] en el método 529: “Determination of explosives and related compounds in drinking water by solid phase extraction and capillary column gas chromatography/mass spectrometry” (GC/MS). Una de las diferencias más importantes entre el método utilizado y el método 529 de EPA es la ausencia de un sistema de inyección con temperatura de vaporización programada (PTV), que permite modificar la temperatura durante la inyección de la muestra e inyectar volúmenes superiores a los 10 μL en el cuerpo de PTV, con el fin de concentrar las muestras al ingresar a la columna [29]. La otra diferencia importante es la temperatura inicial del inyector; el método 529 de EPA [17] recomienda una temperatura del inyector baja debido a que algunas de las sustancias explosivas son muy termolábiles, pero en el equipo utilizado la temperatura mínima recomendada es de 150 ºC, ya que normalmente opera en un rango de 200 ºC a 300 ºC. Por otra parte, es importante mencionar que, además del método en modo splitless recomendado, se probó el método con las mismas condiciones, pero en modo splitless pulsado, el cual había dado buenos resultados con otro tipo de analitos; sin embargo, en el caso de los explosivos en el estudio no hubo mejora alguna, por lo tanto, se decidió continuar con el método en modo splitless.
El mismo método se utilizó para el análisis mediante el GC-NPD; no obstante, se ajustaron las condiciones de algunos parámetros con expertos en toxicología. En este método sí se utilizó en modo splitless pulsado, para empujar los analitos a la columna y evitar su permanencia en el liner. Las condiciones utilizadas se detallan a continuación.
Condiciones instrumentales de análisis
En los siguientes cuadros se detallan las condiciones instrumentales de análisis mediante el GC-MS y el GC-NDP.
Cuadro 1. Condiciones instrumentales de análisis mediante el GC-MS
|
Condiciones del inyector |
Condiciones del horno |
Columna capilar |
Detector MS |
|
Modo: splitless Volumen de inyección: 1,00 μL Temperatura inicial: 150 °C Presión: 12,691 psi Flujo total: 64,542 mL/min Purga del septum: 3 mL/min Gas auxiliar: 16,4 mL/min después de 4 min Purga de flujo al split vent: 60 mL/min a los 2 min Tipo de gas: helio, grado UAP Tiempo total: 29,0 min |
Temperatura máxima: 325 ºC Tiempo de equilibrio: 1 min Programación de temperatura: 45 ºC por 3,3 min, luego una rampa de 10 °C/min hasta 210 °C por 0 min y una rampa de 20 °C/min hasta 250 °C por 7,2 min Tiempo de corrida: 29 min |
Agilent 19091S-433HP-5MS 5% Phenyl Methyl Silox 30 m x 250 μm x 0,25 μL Flujo: 1,54 mL/min Presión inicial nominal: 12,691 psi Velocidad promedio: 45,138 cm/s Tiempo de espera: 1,32 min |
Retraso de disolvente: 4,50 min Gain Factor: 1 Voltaje EM resultante: según archivo de sintonización autotune Rango de detección de masa: de 42 a 420 uma Umbral: 150 MS cuadrupolo: 150 °C MS fuente: 230 °C Modo: scan |
Validación de los métodos establecidos por GC-MS y GC-NPD
Los parámetros de desempeño evaluados para los métodos cromatográficos establecidos fueron el límite de detección, así como a variabilidad en el tiempo de retención para cada analito, en términos de repetibilidad y precisión intermediaa. La estimación del límite de detección instrumental en los métodos por GC-MS y GC-NPD para los analitos evaluados, se realizó mediante el método de Miller & Miller, según la siguiente ecuación [24]:
y = yB + 3 SB (Ecuación 1)
Donde
y = concentración que genera la señal en el instrumento
yB = la ordenada en el origen de la recta de calibrado
SB = la desviación estándar de la recta de calibrado
Cuadro 2. Condiciones instrumentales de análisis mediante el GC-NPD
|
Condiciones del inyector |
Condiciones del horno |
Columna capilar 1 |
Columna capilar 2 |
Detector NPD1 B: |
Detector NPD2 A: |
|
Modo: splitless pulsado Volumen de inyección: 2,00 μL Temperatura inicial: 150 °C Presión: 18,11 psi Presión pulsada: 25,00 psi Tiempo pulsado: 0,50 min Flujo de purga: 50,0 mL/min Tiempo de purga: 1,50 min Flujo total: 56,1 mL/min Gas auxiliar: encendido Corte de solvente: 5,00 min Tipo de gas: helio |
Temperatura inicial: 55 °C Tiempo inicial: 3,30 min Temperatura máxima: 310 ºC Tiempo de equilibrio: 1,00 min Programación de temperatura: rampa de 7 °C/min hasta 210°C por 0 min y rampa de 20 °C/min hasta 250 °C por 5,56 min Temperatura posterior a la corrida: 70 ºC Tiempo posterior a la corrida: 0 min Tiempo de corrida 33 min |
Modelo: Agilent 19091S-433HP-5MS 5 % Phenyl Methyl Siloxane 30 m x 250 μm x 0,25 μL Modo: flujo constante Temperatura máxima: 325 ºC Flujo inicial: 1,7 mL/min Presión inicial nominal: 18,11 psi Velocidad promedio: 37,0 cm/s |
Modelo: JW 122-0732 (14 % cianopropil-fenil)-metilpolisiloxano 30 m x 250 μm x 0,25 μL Modo: flujo constante Temperatura máxima: 300 ºC Flujo inicial: 1,7 mL/min Presión inicial nominal: 18,11 psi Velocidad promedio: 37,0 cm/s |
Temperatura: 320 ºC Flujo de hidrógeno: 3,0 mL/min Flujo de aire: 60,0 mL/min Modo: columna constante con cambio de flujo Flujo combinado: 7,0 mL/min Cambio de flujo: encendido Gas auxiliar: nitrógeno (5,3 mL/min) Tiempo de equilibrio: 5,00 min |
Temperatura: 320 ºC Flujo de hidrogeno: 3,0 mL/min Flujo de aire: 60,0 mL/min Modo: columna constante con cambio de flujo Flujo combinado: 7,0 mL/min Cambio de flujo: encendido Gas auxiliar: nitrógeno (5,3 mL/min) Tiempo de equilibrio: 5,00 min |
Para determinar el límite de detección, en tres fechas diferentes, se prepararon y se inyectaron los patrones para establecer las curvas de calibración de cada analito (durante 6 meses para el GC-MS y 11 meses para el GC-NPD). En cada fecha se inyectó la curva por quintuplicado a diferentes concentraciones, dependiendo del analito (ver cuadros 3 y 4). El cálculo del límite de detección se realizó con una hoja de cálculo en el programa Microsoft Excel®88, con base en el método de Miller & Miller [24].
Para la comprobación del límite de detección y de acuerdo a lo recomendado por Skoog [25], se verificó la relación señal/ruido en el calibrador de concentración más cercana a la concentración estimada del límite de detección. Se determinó la variabilidad en el tiempo de retención para cada analito, en términos de repetibilidad y precisión intermedia. Para determinar la repetibilidad en tres diferentes fechas, se inyectó la curva de calibración de cada analito (durante 6 meses para el GC-MS y 11 meses para el GC-NPD), en cada fecha por quintuplicado, a diferentes concentraciones dependiendo del analito (cuadros 3 y 4). Se realizó un promedio de los tiempos de retención de cada analito; se determinaron las desviaciones estándar y los coeficientes de variación de los tiempos de retención para cada analito en cada una de las tres fechas. Finalmente la estimación de la precisión intermedia, se evaluó como el límite de repetibilidad entre las tres corridas, utilizando ecuaciones (2 y 3) que consideraran la desviación estándar de la repetibilidad y las desviaciones estándares de los tres eventos (tres corridas).
Sr = (S12 + S22 + S32 / 3)1/2 (Ecuación 2)
Límite de Sr = 2,8 x Sr (Ecuación 3)
Donde:
Sr = Desviación estándar de la repetibilidad
S1 = Desviación estándar en el primer evento
S2 = Desviación estándar en el segundo evento
S3 = Desviación estándar en el tercer evento
Cuadro 3. Concentraciones y número de réplicas de cada analito para el cálculo del límite de detección en el GC-MS, mediante el método de Miller & Miller
|
Analito |
Cantidad de réplicas totales de cada analito en los tres eventos |
Concentraciones para la curva de calibración (mg/L) |
|
1,3,5-trinitrotolueno |
15 |
75, 100, 125, 150 |
|
2,4,6-trinitrotolueno |
15 |
4, 6, 8, 10, 15 |
|
2,4-dinitrotolueno |
15 |
4, 6, 8, 10, 15 |
|
2,6-dinitrotolueno |
15 |
4, 6, 8, 10, 15 |
|
2-nitrotolueno |
15 |
4, 6, 8, 10, 15 |
|
3-nitrotolueno |
15 |
4, 6, 8, 10, 15 |
|
4-nitrotolueno |
15 |
4, 6, 8, 10, 15 |
|
Difenilamina |
15 |
4, 6, 8, 10, 15 |
|
Etilcentralita |
15 |
4, 6, 8, 10, 15 |
|
Nitroglicerina |
15 |
75, 100, 125, 150 |
|
PETN |
15 |
75, 100, 125, 150 |
|
RDX |
15 |
75, 100, 125, 150 |
|
Tetril |
15 |
75, 100, 125, 150 |
Detonaciones
Se realizaron 6 detonaciones de pequeñas cantidades de material explosivo de diferente naturaleza, bajo condiciones controladas. A los Artefactos Explosivos Improvisados (IED) que se utilizaron en las pruebas de campo, se les asignaron las identificaciones internas EXPLO-1-PE, EXPLO-2-PE, EXPLO-3-PE, EXPLO-4-PE, EXPLO-4-PE, EXPLO-5-I y EXPLO-6-PE
Cuadro 4. Concentraciones y número de réplicas de cada analito para el cálculo del límite de detección en el GC-NPD, mediante el método de Miller & Miller
|
Analito |
Cantidad de réplicas totales de cada analito en los tres eventos |
Concentraciones para la curva de calibración (mg/L) |
|
2,4,6-trinitrotolueno |
15 |
4, 6, 8, 10, 15 |
|
2,4-dinitrotolueno |
15 |
4, 6, 8, 10, 15 |
|
2,6-dinitrotolueno |
15 |
4, 6, 8, 10, 15 |
|
2-nitrotolueno |
15 |
4, 6, 8, 10, 15 |
|
3-nitrotolueno |
15 |
4, 6, 8, 10, 15 |
|
4-nitrotolueno |
15 |
4, 6, 8, 10, 15 |
|
Difenilamina |
15 |
4, 6, 8, 10, 15 |
|
Etilcentralita |
15 |
4, 6, 8, 10, 15 |
|
Nitroglicerina |
15 |
4, 6, 8, 10, 15 |
|
RDX |
15 |
4, 6, 8, 10, 15 |
|
Tetril |
15 |
4, 6, 8, 10, 15 |
Recolección de los indicios post-explosión:
En la recolección de la muestra se siguió lo recomendado por Akhanan [1] y Thurman [26]. El material recolectado se almacenó en una bolsa de papel kraft debidamente identificada, para ser trasladado de esta forma al laboratorio para su posterior análisis.
Análisis cromatográfico de las muestras por GC-MS y GC-NPD
Se realizaron análisis de cada una de las muestras cuestionadas, empleando controles positivos y negativos, así: a) al iniciar la secuencia de inyección, se colocaron al menos tres blancos instrumentales de acetato de etilo para verificar la limpieza del sistema; b) se colocaron dos blancos instrumentales de acetato de etilo previamente a cada muestra cuestionada, a cada control negativo y a cada control positivo; c) se les dio prioridad de inyección a los controles negativos, seguidamente a las muestras cuestionadas y por último a los controles positivos, para que estos no contaminaran las muestras cuestionadas ni los controles negativos, y d) se colocaron al menos tres blancos instrumentales de acetato de etilo al final de la secuencia para verificar la limpieza del instrumento.
Resultados
Por GC-MS
En la figura 1 se muestra la separación cromatográfica obtenida de la mezcla GC-MS-1 a una concentración de 10 mg/L, compuesta por 2,4-dinitrotolueno, 2,6-dinitrotolueno, nitroglicerina, etilcentralita, difenilamina, N-nitrosodifenilamina, 4-amino-2,6-dinitrotolueno y 2-amino-4,6-dinitrotolueno. La separación cromatográfica de la mezcla GC-MS-1 presentó una distribución de picos cromatográficos bien resueltos de todos los componentes, que aparecieron primero 13 minutos después de dar inicio la corrida y continuaron apareciendo hasta antes de completar los 21 minutos, ubicándose hacia la parte media de la duración total de la corrida cromatográfica. Como se muestra en la figura 1, se logró una separación cromatográfica de cada uno de los analitos incluidos en la mezcla GC-MS-1, según sus tiempos de retención, excepto de la difenilamina y la N-nitrosodifenilamina porque ambas sustancias coeluyen y las bibliotecas espectrales utilizadas identifican la señal obtenida únicamente como difenilamina. Con base en estos resultados se descartó la N-nitrosodifenilamina como analito. La señal con alta abundancia entre los 13,5 y los 14,0 minutos corresponde a la nitroglicerina; no obstante se recomienda la búsqueda por iones para confirmar la presencia de este compuesto.
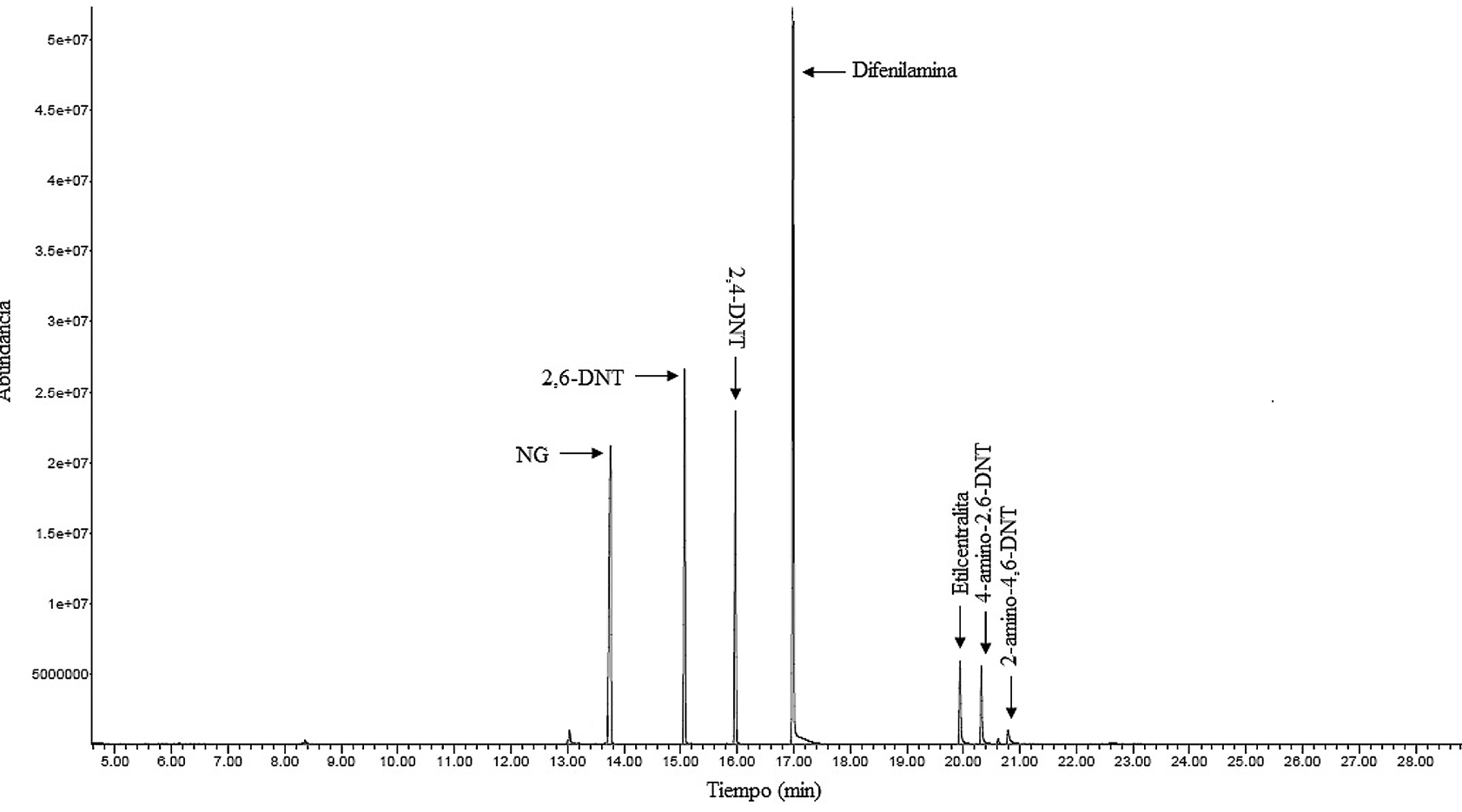
Figura 1. Cromatograma de iones totales de la mezcla GC-MS-1 a 10 mg/L, en cada componente
La figura 2 muestra la separación cromatográfica obtenida para los analitos que componían la mezcla GC-MS-2. Es evidente que fue posible resolver adecuadamente las señales para los diferentes analitos, por lo tanto, la separación cromatográfica de la mezcla GC-MS-2 presentó una distribución de picos cromatográficos adecuadamente resueltos de todos los componentes, que aparecieron primero a los 10 minutos después de iniciar la corrida, y continuaron apareciendo hasta antes de completar los 22 minutos, ubicándose en la parte media de la duración total de la corrida cromatográfica. El comportamiento de la separación cromatográfica de la mezcla GC-MS-2 fue muy similar al de la mezcla GC-MS-1. Los tiempos (min.) de retención estimados de los analitos de la mezcla GC-MS-1 en orden de elución fueron nitroglicerina (13,807), 2,6 dinitrotolueno (15,137), 2,4 dinitrotolueno (16,037), difenilamina (17,059), etilcentralita (20,013), 2-amino-4,6-dinitrotolueno (20,943) y 4-amino-2,6-dinitrotolueno (20,488). En el caso de la mezcla GC-MS-2, los tiempos (min) de retención de los analitos en orden de elución fueron 2-nitrotulueno (10,939), 3-nitrotulueno (11,432), 4-nitrotulueno (11,755) y 2,4,6-trinitrotolueno (18,023).
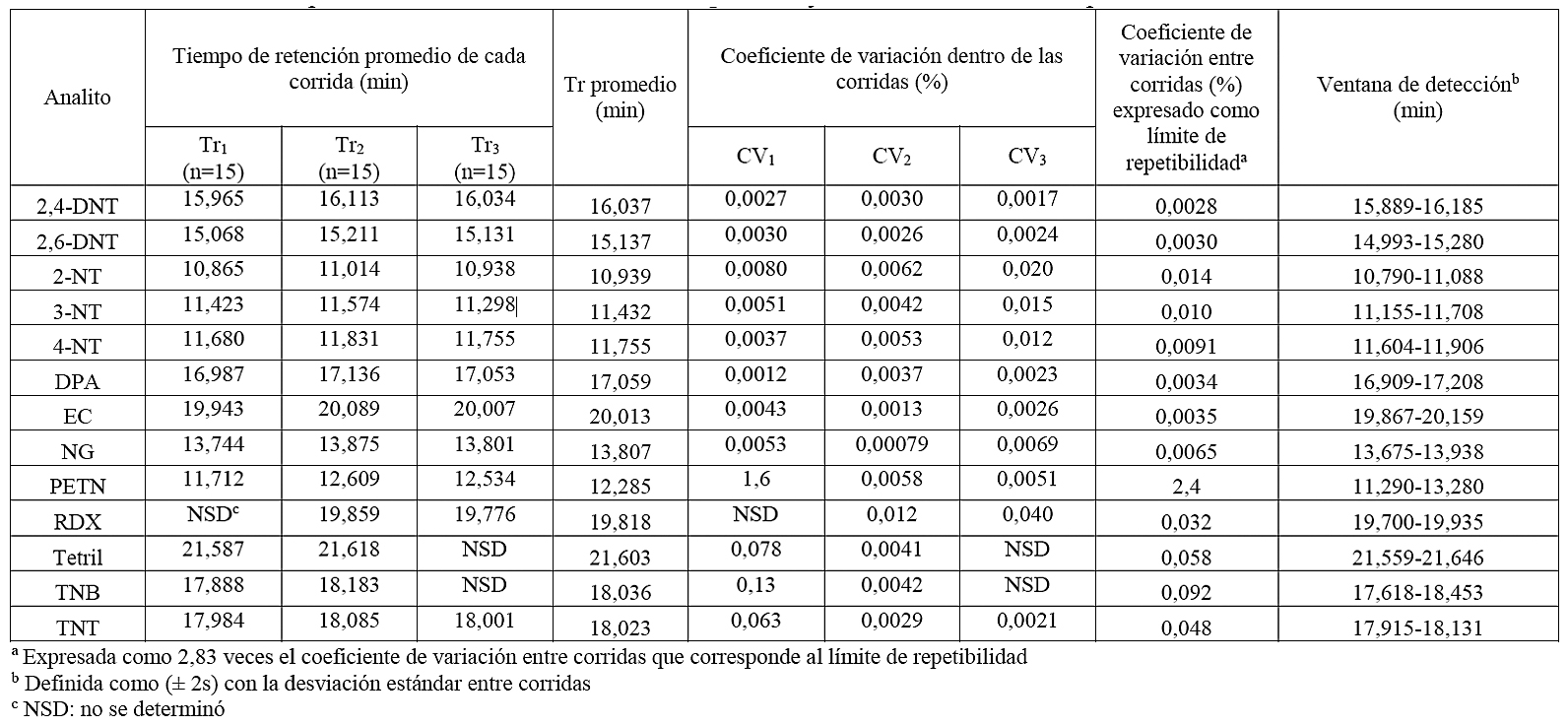
Según los datos de tiempo de retención, se tiene que el analito RDX exhibió una pecualiaridad que no se manifestó en los otros analitos, debido a la aparición de una señal constante a 19,8 minutos. Esto pudo ser debido a que el mecanismo de descomposición térmica del RDX no está del todo clarificado. Estudios de Underill y Bardon [27] indican que inicialmente debe ocurrir una pérdida de HNO2 y la formación de un doble enlace entre el carbono y el nitrógeno. Los límites de detección (mg/L, valor de la concentración del analito en el extracto o la cantidad de nanogramos de analito inyectados en un µL de extracto) estimados para los analitos en estudio por GC-MS, calculados bajo el método de Miller & Miller fueron 1,3,5-trinitrobenceno (58), 2,4,6-trinitrotolueno (0,99), 2,4-dinitrotolueno (1,31), 2,6 dinitrotolueno (0,86), 2-nitrotolueno (0,32), 3-nitrolueno (0,35), 4-nitrotolueno (0,61), difenilamina (3,3), etilcentralita (1,8), nitroglicerina (2,4), PETN (5,7), RDX (92) y tetril (14).
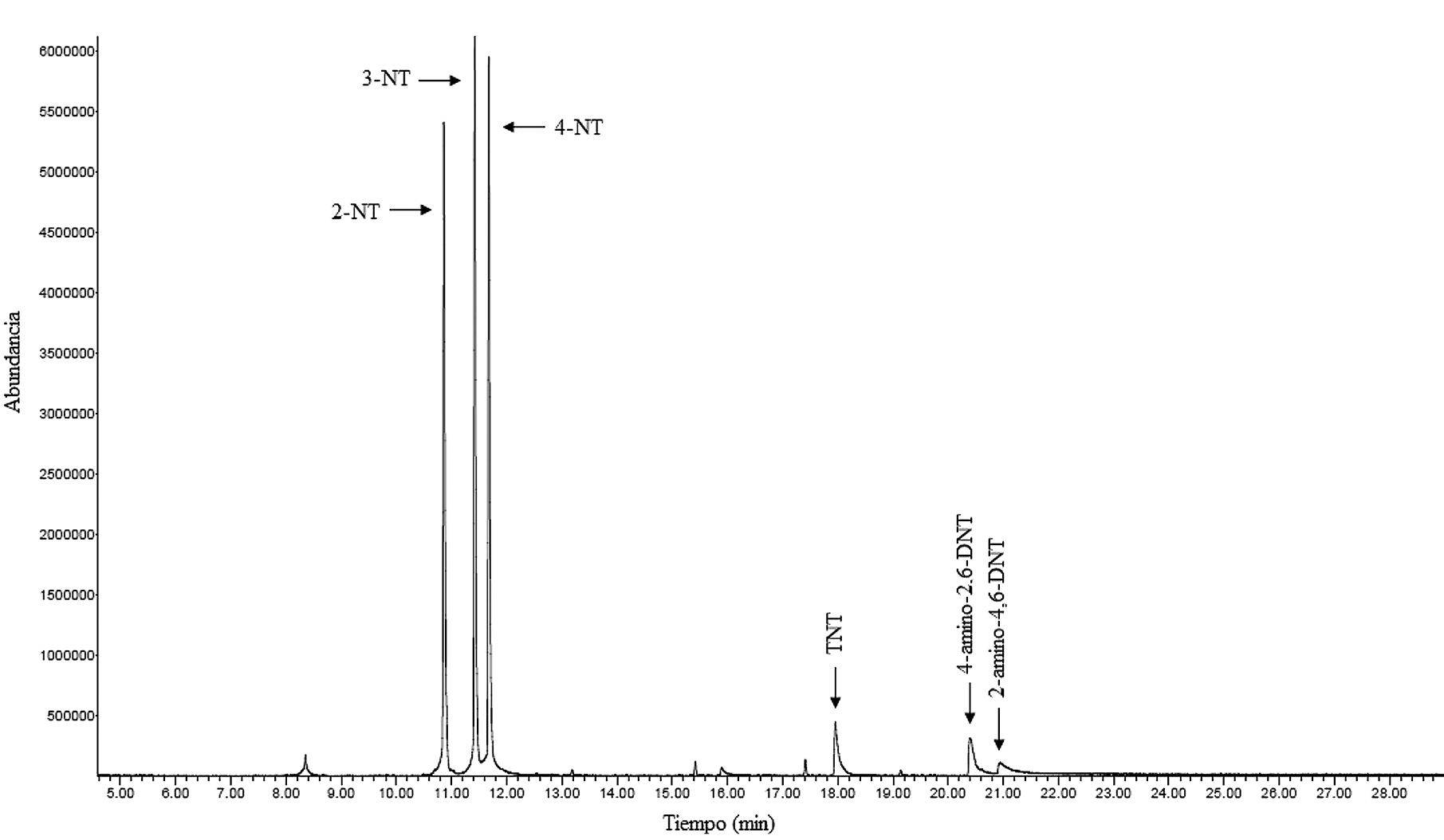
Figura 2. Cromatograma de iones totales de la mezcla GC-MS-2 a 10 mg/L, en cada componente.
La repetibilidad del tiempo de retención se determinó midiendo la variabilidad de los tiempos de retención dentro de cada una de las tres corridas que se realizaron, para cada una de las cuales se determinaron las desviaciones estándar y los coeficientes de variación de los tiempos de retención de cada analito. La precisión del tiempo de retención de cada analito dentro de una misma corrida fue bastante buena, y se obtuvieron coeficientes de variación entre 0,0012 % y 1,6 % para todos los analitos, como se aprecia en el anexo I. Esto contrasta con la variabilidad entre corridas, pues se obtuvieron coeficientes de variación entre 0,0028 % y 2,4 % (expresados como límites de repetibilidad). Las precisiones alcanzadas en estas condiciones se utilizaron para establecer las ventanas de detección ampliadas como ± 2 veces la desviación estándar entre las tres corridas, tal como se muestra en el anexo I. Estos resultados satisfacen lo que tradicionalmente es aceptado como variabilidad máxima en este tipo de determinaciones (± 2 % o 0,1 min) [12] considerando que durante el período en estudio se realizaron cambios de columna, cortes en columna y procesos de mantenimiento general. Con respecto a la precisión intermedia, esta mide la precisión de un método realizando un análisis sobre una misma muestra en el mismo laboratorio y con el mismo método, pero con distinto analista o en diferentes corridas con períodos distantes entre ellas. En este caso, debido a que no se contó con datos generados por otros analistas, se estimó la precisión intermedia como un límite de repetibilidad entre las tres corridas realizadas.
Por GC-NPD
Respecto a los resultados obtenidos mediante el método GC-NPD, en la figura 3 se muestran los cronogramas respectivos. Según la figura, el cromatograma con señales obtenidas con el detector NPD1 B, acoplado a una columna capilar J&W 122-0732 con una polaridad de media a baja, presentó una mayor resolución de los picos cromatográficos y en mayor abundancia, pero a la vez produjo dos traslapes: el primero entre el 2,4-dinitrotolueno y la nitroglicerina, y luego entre la etilcentralita y el TNT; el segundo traslape fue de mayor importancia. En el caso de la respuesta cromatográfica para el detector NPD2 A, acoplado a una columna capilar HP-5MS no polar, presentó una mejor separación de los picos cromatográficos; no existieron traslapes entre los analitos evaluados, pero también presentó menor abundancia en comparación con la columna con mayor polaridad. Los tiempos de retención obtenidos para los analitos de la figura 3 se detallan en el cuadro 5.
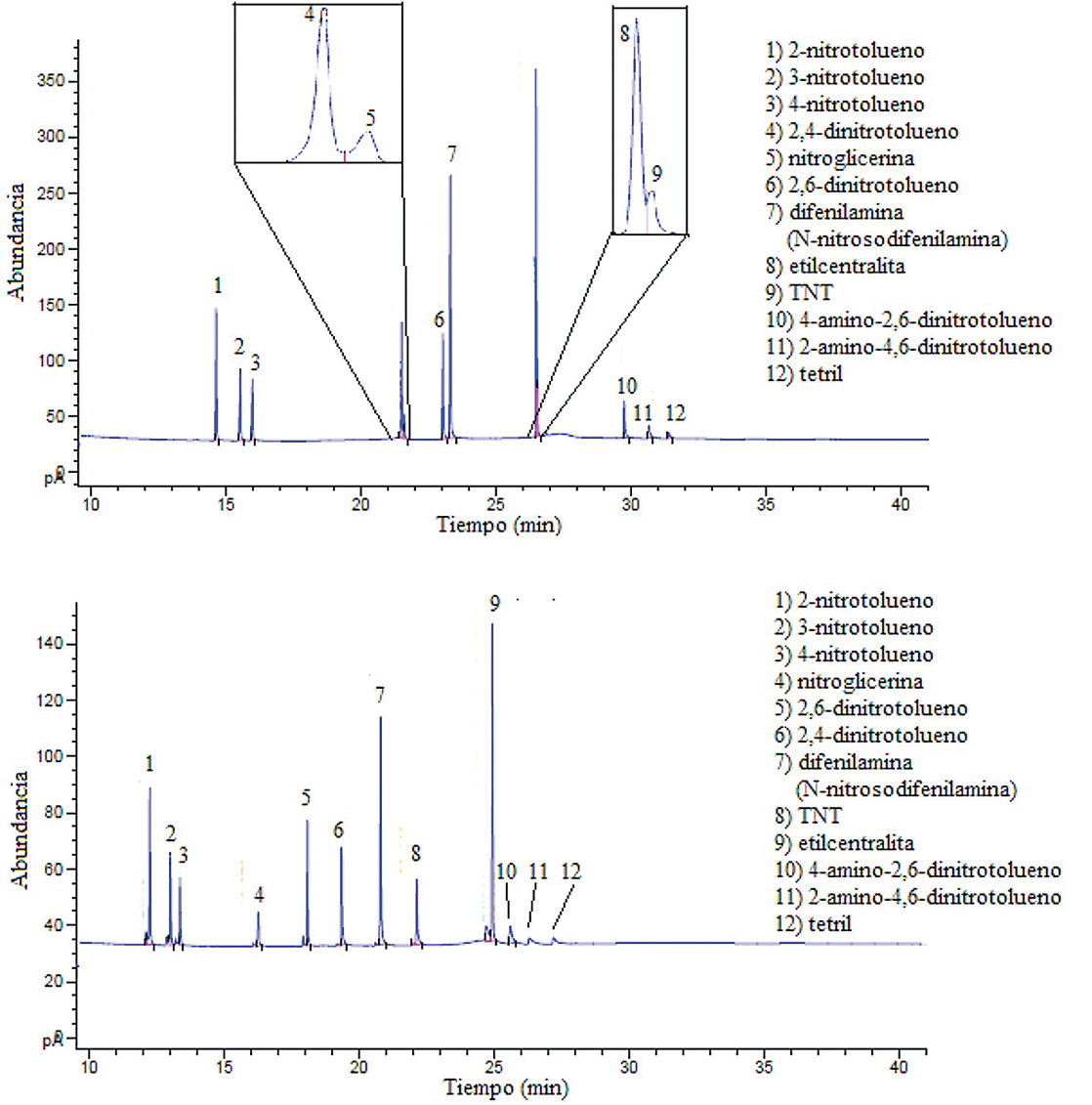
Figura 3 Arriba: Cromatograma para una mezcla de analitos a 10 mg/L en el detector NPD1 B, con una columna capilar J&W DB-1701. Abajo: Cromatograma para una mezcla de analitos a 10 mg/L en el detector NPD2 A, con una columna capilar HP-5MS.
En el cuadro 6 se muestran los límites de detección estimados para los analitos en estudio, calculados bajo el método de Miller & Miller, para el procedimiento por GC-NPD.
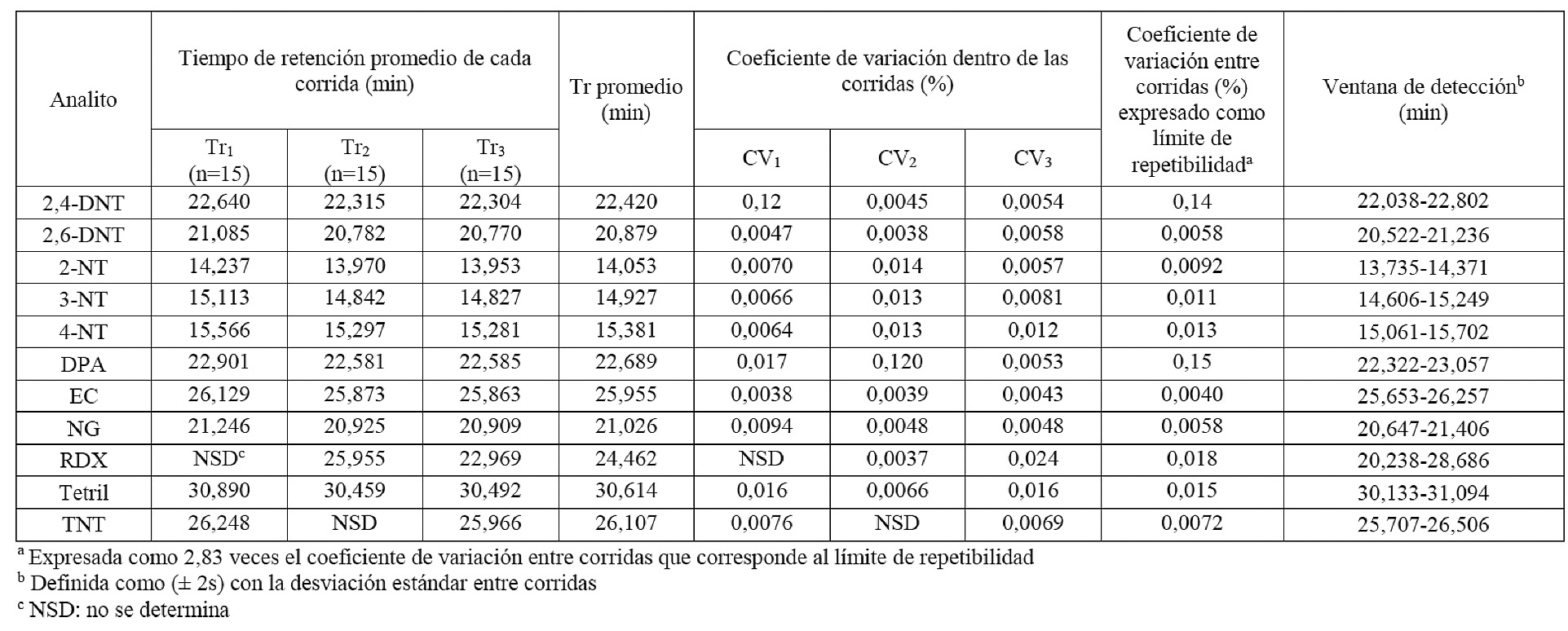
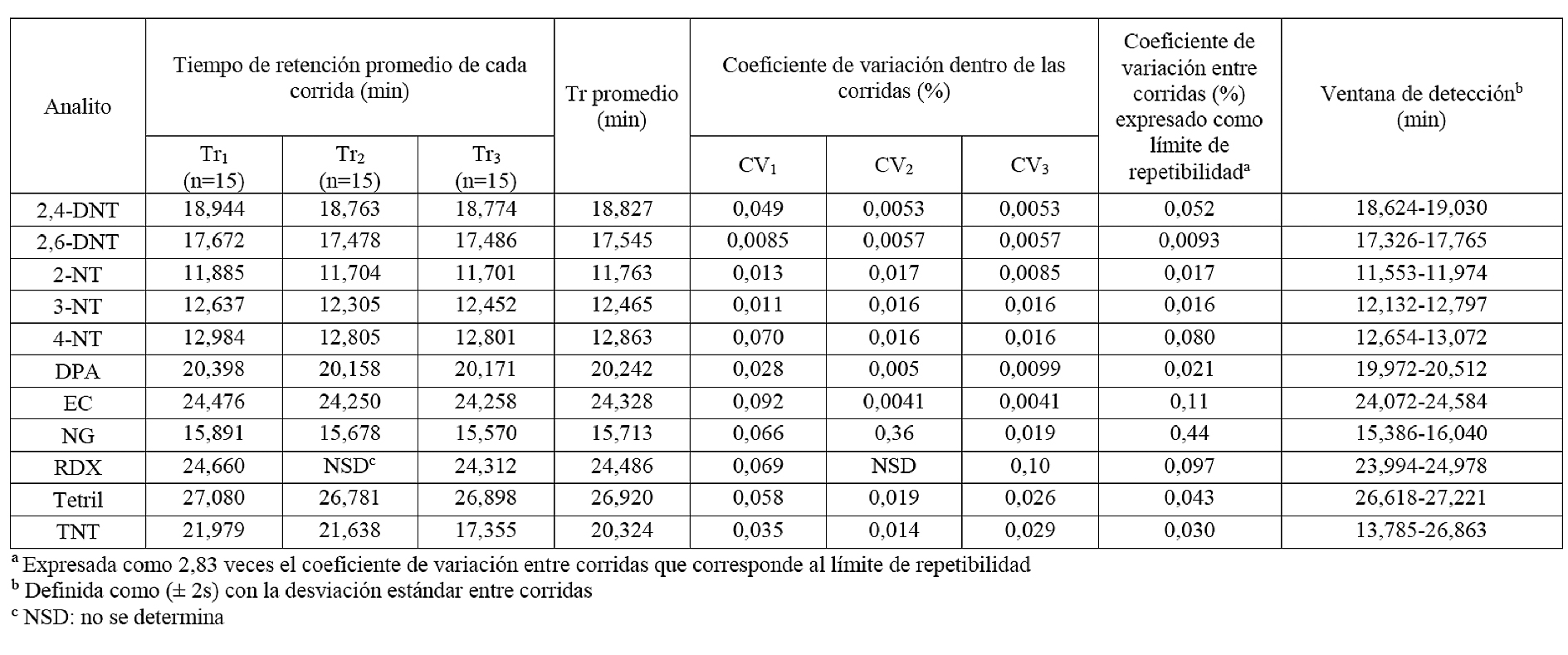
Según el cuadro 6, si se comparan los resultados de los límites de detección instrumental por el método establecido para los patrones de referencia evaluados por GC-MS, se observa que los valores de los límites de detección son muy diferentes entre el GC-MS y el GC-NPD. Por el método utilizado se obtuvieron límites de detección más altos con el detector MS que con el NPD, que presentó límites de detección mucho menores debido a su alta selectividad. En los cuadros II y III de la sección de anexos, se muestran los resultados obtenidos para la repetibilidad de los analitos evaluados mediante los dos sistemas cromatográficos del GC-NPD. Al igual que en el GC-MS, las variabilidades alcanzadas con el método propuesto en el detector GC-NPD demostraron la capacidad que tienen los métodos utilizados para mantener unos resultados concordantes y próximos entre sí, con referencia a su valor medio, bajo las mismas condiciones de laboratorio y método y por el mismo analista, en una misma corrida o entre corridas.
Cuadro 5. Tiempos de retención promedio estimados por el método establecido para los analitos evaluados por GC-NPD, con doble sistema de columna cromatográfica
|
Analito |
Tiempo de retención |
Tiempo de retención |
|
2-nitrotolueno |
14,053 |
11,781 |
|
3-nitrotolueno |
14,927 |
12,465 |
|
4-nitrotolueno |
15,381 |
12,864 |
|
2,6 dinitrotolueno |
20,897 |
17,545 |
|
Nitroglicerina |
21,026 |
15,765 |
|
2,4 dinitrotolueno |
22,240 |
18,827 |
|
Difenilamina |
22,689 |
20,243 |
|
RDX |
24,462 |
24,486 |
|
Etilcentralita |
25,955, |
24,328 |
|
2,4,6 trinitrotolueno |
26,107 |
21,781 |
|
Tetril |
30,614 |
26,920 |
|
4-amino-2,6 dinitrotolueno |
29,750 |
No se determinó. |
|
2-amino-4,6 dinitrotolueno |
30,695 |
No se determinó. |
Cuadro 6. Límites de detección instrumental por el método establecido en GC-NPD para los analitos evaluados, en ambos sistemas cromatográficos
|
Analito |
Límite de detección estimadoa,b / mg/L (entre paréntesis la cantidad de ng en columna) |
|
|
Sistema J&W DB-1701 NPD1 B |
Sistema HP-5MS-NPD2 A |
|
|
2,4,6-trinitrotolueno |
0,05 (0,10) |
0,81 (1,62) |
|
2,4-dinitrotolueno |
0,03 (0,06) |
0,01 (0,02) |
|
2,6-dinitrotolueno |
0,04 (0,08) |
0,04 (0,08) |
|
2-nitrotolueno |
0,38 (0,76) |
0,03 (0,06) |
|
3-nitrotolueno |
0,06 (0,12) |
0,05 (0,10) |
|
4-nitrotolueno |
0,09 (0,18) |
0,05 (0,10) |
|
Difenilamina |
0,12 (0,24) |
0,01 (0,02) |
|
Etilcentralita |
0,02 (0,04) |
0,02 (0,04) |
|
Nitroglicerina |
4,00 (8) |
4,00 (8) |
|
RDX |
0,17 (1,34) |
0,04 (0,08) |
|
Tetril |
3,21 (6,42) |
3,06 (6,12) |
a Corresponde al valor de la concentración del analito en el extracto o la cantidad de nanogramos de analito inyectados en2µL de extracto.
b Los límites de detección se estimaron según Miller &Miller, excepto para la NG que se estableció según la relación señal/ruido.
Análisis de muestras reales
Para evaluar el desempeño del método establecido tanto en el proceso de extracción como en la identificación de los componentes explosivos en los indicios post-explosión, se procedió a realizar seis detonaciones de artefactos explosivos improvisados. Para la verificación de la eficiencia en la identificación de los explosivos por GC-NPD, se seleccionaron solo algunas de las muestras que se habían utilizado por GC-MS. En el cuadro 10 se presentan los resultados de dichos análisis (GC-MS) para el artefacto EXPLO-3-PE. El resto de los resultados se muestran en el anexo IV. Para el artefacto EXPLO-1-PE, no se detectó el RDX, al igual que por GC-NPD, lo cual, según Borusiewiez [28], podría estar relacionado con la eficiencia de la detonación, de manera que la cantidad de los residuos de los explosivos sin detonar que dejó la explosión del artefacto estuviera por debajo de los niveles de detección, dependiendo de una serie de parámetros como la forma de la carga, las propiedades y la posición del detonador. No obstante, la condición de mejores límites de detección encontrados por GC-NPD permitió la identificación de RDX en una de las muestras post-explosión del artefacto EXPLO-2-PE (en los dos detectores), mientras que en otra de las muestras solo se detectó RDX en la columna conectada al detector NPD2 A, cuyo límite de detección de RDX fue de un orden de magnitud menor con respecto al del sistema cromatográfico que tiene el detector NPD1 B.
Cuadro 7. Resultados del análisis de indicios post-explosión por GC-MS
|
Artefacto |
Sustancia buscada |
Sustancia encontrada |
Tiempo de retención/(min) |
Quality match |
|
EXPLO-3-PE |
PETN TNT |
2,4-DNT |
15,851 |
97 |
|
TNT |
17,880 |
90 |
||
|
4-amino-2,6-DNT |
20,342 |
97 |
||
|
2,6-DNT |
14,944 |
76 |
||
|
2,6-DNT |
14,949 |
98 |
||
|
2,4-DNT |
15,851 |
98 |
||
|
TNT |
17,964 |
90 |
||
|
4-amino-2,6-DNT |
20,324 |
99 |
||
|
TNT |
17,866 |
90 |
||
|
2,6-DNT |
14,944 |
90 |
||
|
2,4-DNT |
15,851 |
91 |
||
|
TNT |
17,880 |
90 |
||
|
4-amino-2,6-DNT |
20,355 |
97 |
||
|
2,6-DNT |
14,949 |
98 |
||
|
2,4-DNT |
15,841 |
96 |
||
|
TNT |
17,858 |
91 |
||
|
2,6-DNT |
14,949 |
90 |
||
|
2,4-DNT |
15,851 |
98 |
||
|
TNT |
17,897 |
90 |
||
|
2,6-DNT |
14,975 |
90 |
||
|
2,4-DNT |
15,886 |
96 |
||
|
TNT |
17,827 |
87 |
and: no detectable
En una de las muestras analizadas por GC-NPD derivadas del explosivo EXPLO-6-PE se verificó la existencia de TNT, cuando en realidad este artefacto explosivo no contenía dicho compuesto; fue la segunda ocasión en este estudio en que se observó una contaminación con TNT, que estuvo como componente mayoritario en las cargas de tres de los seis artefactos explosivos utilizados en esta investigación. Respecto a los resultados obtenidos (algunos se muestran en el cuadro 7 y el anexo IV, respectivamente), se tiene que la totalidad de los análisis realizados indican que fue posible la identificación definitiva de al menos uno de los componentes explosivos en el 66,7 % de los artefactos explosivos detonados (EXPLO-3-PE. EXPLO-4-PE, EXPLO-5-PE y EXPLO-6-PE), tanto por GC-MS como por GC-NPD (doble separación) y TLC. En un 16,7 % de los casos (EXPLO-2-PE) se pudo dar una identificación tentativa, pues solo se contaba con dos resultados de pruebas ortogonales realizadas (GC-NPD con doble sistema de separación y detección), y finalmente en un 16,7 % de las pruebas no fue posible detectar en los restos post-explosión el explosivo utilizado. De esta manera, en un 83 % de los experimentos se demostró la eficacia de la batería analítica utilizada en este caso, como conjunto de pruebas que, con una dependencia de la técnica, pueden sostener un resultado de identificación química por sí mismas de forma independiente, o bien complementarse entre ellas para obtener una categorización de la sustancia explosiva.
Conclusiones
La separación cromatográfica (procedimiento por GC-MS) de la mezcla GC-MS-1 presentó una distribución de picos cromatográficos bien resueltos para todos los componentes; su aparición se inició a los 13 minutos y continuó hasta antes de completar los 21 minutos; los picos se ubicaron hacia la parte media de la duración total de la corrida cromatográfica.
Mediante el método por GC-MS se logró una separación cromatográfica de cada uno de los analitos incluidos en la mezcla GC-MS-1, según sus tiempos de retención, excepto de la difenilamina y la N-nitrosodifenilamina, porque ambas sustancias coeluyen y las bibliotecas espectrales utilizadas identifican la señal obtenida únicamente como difenilamina.
Los límites de detección instrumental por el método establecido para los patrones de referencia evaluados por GC-MS oscilan en el rango de 0,32 mg/L a 58 mg/L. El menor límite de detección corresponde al analito 2-nitrotolueno y el mayor límite de detección a RDX.
Mediante el método por GC-MS se obtuvo una precisión del tiempo de retención bastante buena para cada analito dentro de una misma corrida y los coeficientes de variación de los analitos se ubicaron en un rango de 0,0012% a 1,6%”.
Respecto al método por GC-NPD, el cronograma con señales obtenidas con el detector NPD1 B, acoplado a una columna capilar J&W 122-0732 con una polaridad de media a baja, presentó mayor resolución de los picos cromatográficos y mayor abundancia de estos con respecto al detector NDP2 A.
Los valores de los límites de detección fueron muy diferentes entre el GC-MS y el GC-NPD. Se obtuvieron límites de detección más altos con el MS que con el NPD, que presentó límites de detección mucho menores debido a la alta selectividad de este detector.
De forma similar que por el GC-MS, la variabilidad alcanzada con el método propuesto por el GC-NPD demostró la capacidad de los métodos utilizados para mantener unos resultados concordantes y próximos entre sí con referencia a su valor medio, bajo las mismas condiciones delaboratorio y método y por el mismo analista, en una misma corrida o entre corridas.
Para el caso particular de la muestra real EXPLO-1-PE, no se detectó RDX (mediante las técnicas GC-NPD y GC-MS), lo cual pudo estar relacionado con la eficiencia de la detonación, de manera que la cantidad de los residuos de los explosivos sin detonar que dejó la explosión del artefacto estuviera por debajo de los niveles de detección.
La condición de mejores límites de detección encontrada en el método por GC-NPD permitió la identificación de RDX en una de las muestras post-explosión del artefacto EXPLO-2-PE (en los dos detectores), mientras que en otra de las muestras solo se detectó RDX en la columna conectada al detector NPD2 A, cuyo límite de detección de RDX fue de un orden de magnitud menor con respecto al del sistema cromatográfico con el detector NPD1 B.
La totalidad de los análisis realizados indican que fue posible la identificación definitiva de al menos uno de los componentes explosivos en el 66,7 % de los artefactos explosivos detonados (EXPLO-3-PE. EXPLO-4-PE, EXPLO-5-PE y EXPLO-6-PE), tanto por GC-MS como por GC-NPD (doble separación).
En un 16,7 % de los casos (EXPLO-2-PE), se pudo realizar una identificación tentativa, pues solo se contaba con dos resultados de pruebas ortogonales (por GC-NPD con doble sistema de separación y detección). En un 16,7 % de las pruebas (por ambos métodos) no fue posible detectar en los restos el explosivo utilizado.
La batería analítica establecida para la determinación de explosivos por GC-MS y GC-NPD, resultó bastante eficaz en el análisis de los indicios post-explosión (83 %), en la identificación de al menos un componente explosivo en las muestras derivadas de la detonación controlada de artefactos explosivos improvisados con cargas detonantes o deflagrantes de composición conocida.
Referencias
[1] J. Akhavan, The Chemistry of Explosives, 2nd ed. London: RSC, 2004, pp. 21-27, 149.
[2] R. Meyer, J. Köhler, A. Homburg, Eds. Explosives, 6th ed. Germany: WILEY-VCH Verlag, 2007, pp. 131-132.
[3] A. Beveridge, Ed., Forensic Investigation of Explosions, 2nd ed., (International Forensic Science and Investigations Series). U.S.A.: CRC Press, 2011, pp. 4-23.
[4] S. Venugopalan, Demystifying explosives: Concepts in high energy materials. Russland: Elsevier, 2014, pp. 1-14.
[5] T. Urbanski, Chemistry and technology of explosives, vol. 1. Poland: Pergamon Press, 1964, pp 1-6, 248-255, 268-285.
[6] P. Warey, Ed., New research on hazardous materials. New York: Nova Science Publishers, Inc., 2007, pp 371-373.
[7] “Explosives analysis in the environment,” in Encyclopedia of Analytical Chemistry, vol. 3, John Wiley & Sons Ltd. U.S.A., 2006, pp. 2402-2403.
[8] M. Peterson, “TNT and 4-amino-2,6-dinitrotoluene influence on germination and early seedling development of tall fescue”, in Elsevier, 93, 1995, pp. 57-58.
[9] A. Hewitt, T. Jenkins, and T. Ranney, “On-site gas chromatographic determination of explosives in soils”, in Field analytical chemistry and technology, vol. 5, 2001, pp. 228-229.
[10] US Environmental Protection Agency, “Method 8515: Colorimetric screening method for trinitrotoluene (TNT) in soil.” http://www.epa.gov/solidwaste/hazard/testmethods/sw846/pdfs/8515.pdf. (Recuperado el 26 de julio de 2015).
[11] US Environmental Protection Agency, “Method 8510: Colorimetric screening procedure for RDX and HMX in soil.” http://www.epa.gov/solidwaste/hazard/testmethods/sw846/pdfs/8510.pdf. (Recuperado el 26 de julio de 2015).
[12] US Environmental Protection Agency, “Method 4050: TNT explosives in soil by inmunoassay.” http://www.epa.gov/solidwaste/hazard/testmethods/sw846/pdfs/8510.pdf. (Recuperado el 26 de julio de 2015).
[13] US Environmental Protection Agency, “Method 4051: Hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX) in soil by inmunoassay.” http://www.epa.gov/solidwaste/hazard/testmethods/sw846/pdfs/8510.pdf. (Recuperado el 26 de julio de 2015).
[14] US Environmental Protection Agency, “Method 8330A: Nitroaromatics and nitramines by high performance liquid chromatography (HPLC).” http://www.epa.gov/solidwaste/hazard/testmethods/sw846/pdfs/8330a.pdf. (Recuperado el 26 de julio de 2015).
[15] US Environmental Protection Agency, “Method 8330B: Nitroaromatics, nitramines, and nitrate esters by high performance liquid chromatography (HPLC).” http://www.epa.gov/solidwaste/hazard/testmethods/pdfs/8330b.pdf. (Recuperado el 26 de julio de 2015).
[16] US Environmental Protection Agency, “Method 8095: Explosives by Gas Chromatography.” http://www.epa.gov/solidwaste/hazard/testmethods/sw846/pdfs/8095.pdf. (Recuperado el 26 de julio de 2015).
[17] US Environmental Protection Agency, “Method 529: Determination of explosives and related compounds in drinking water y solid phase extraction and capillary column gas chromatography/mass spectrometry (GC/MS).” http://www.epa.gov/solidwaste/hazard/testmethods/sw846/pdfs/8095.pdf. (Recuperado el 26 de julio de 2015).
[18] W. Schade, R. Orghici, M. Mordmüller, and U. Willer, Detection of explosives. U.S.A.: Wiley-VCH Verlag GmbH & Co. KGaA, 2012, pp. 195-196.
[19] R. Varga, and P. Ulbrich, “Some experience with trace analysis of post-explosion residues.” Arms Technology, no. 3, 2004, p. 634.
[20] E. B. Byall, “Detection and characterization of explosives and explosive residue: A review,” in 13th INTERPOL Forensic Science Symposium, 2001, pp. 100-106.
[21] D. Royds, S. Lewis, and A. Taylor, “A case study in forensic chemistry: The Bali bombings,” Talanta, no. 67, 2005, pp. 262-268.
[22] R. Garrido, “Uso de la espectrometría de movilidad iónica como técnica de vanguardia en los laboratorios analíticos de rutina”, Servicio de Publicaciones de la Universidad de Córdoba, Argentina, 2012, pp. 25-33.
[23] C. L. Rhykerd, D. W. Hannum, D. W. Murray, and J. E. Parmeter, “Guide for the selection of commercial explosives detection systems for Law enforcement applications”, National Institute of Justice, U.S.A., 1999, pp. 10-20.
[24] J. Miller, Estadística y quimiometría para Química Analítica, 4a ed. España: Pearson Education, 2002, pp. 125-126.
[25] D. Skoog, F. Holler y A. Nieman, Principios de análisis instrumental, 5.a ed.. España: McGraw-Hill, 2003, pp. 103-104.
[26] J. Thurman, Practical Bomb Scene Investigation. Canada: CRC Press, 2006, pp. 187-190.
[27] P. R. Underhill and M. F. Bardon, “Modelling of thermal ignition of RDX/HTPB based PBXS”, Royal Millitary Coll of Canada, Canada, 1996, pp. 50-57.
[28] R. Borusiewicz, G. Zadora, and J. Zieba-Palus, “Chemical analysis of post explosion samples obtained as a result of model field experiments,” Talanta, no. 116, 2013, pp. 630-636.
[29] E. Stashenko y J. Martínez, “GC y GC-MS: configuración del equipo versus aplicaciones”, Scientia Chromatographica, n.o 2, 2010, pp. 49-50
[30] Wada Laboratory Committee, “Identification criteria for qualitative assays incorporating column chromatography and mass spectrometry”, Wada Technical Document – TD2010IDCR, version number: 1.0, approved by: WADA Executive Committee, approval date: 8 May, 2010, effective date: 1 September, 2010.
ANEXO I. Estudio de la repetibilidad en la determinación de explosivos y sustancias relacionadas por GC-MS
ANEXO II. Estudio de la repetibilidad en la determinación de los explosivos y las sustancias relacionadas con el sistema J&W DB-1701-NPD1 B del GC-NPD
ANEXO III. Estudio de la repetibilidad en la determinación de explosivos y sustancias relacionadas con el sistema HP-5MS-NPD2 A del GC-NPD
ANEXO IV. Resultados del análisis de indicios post-explosión por GC-MS.
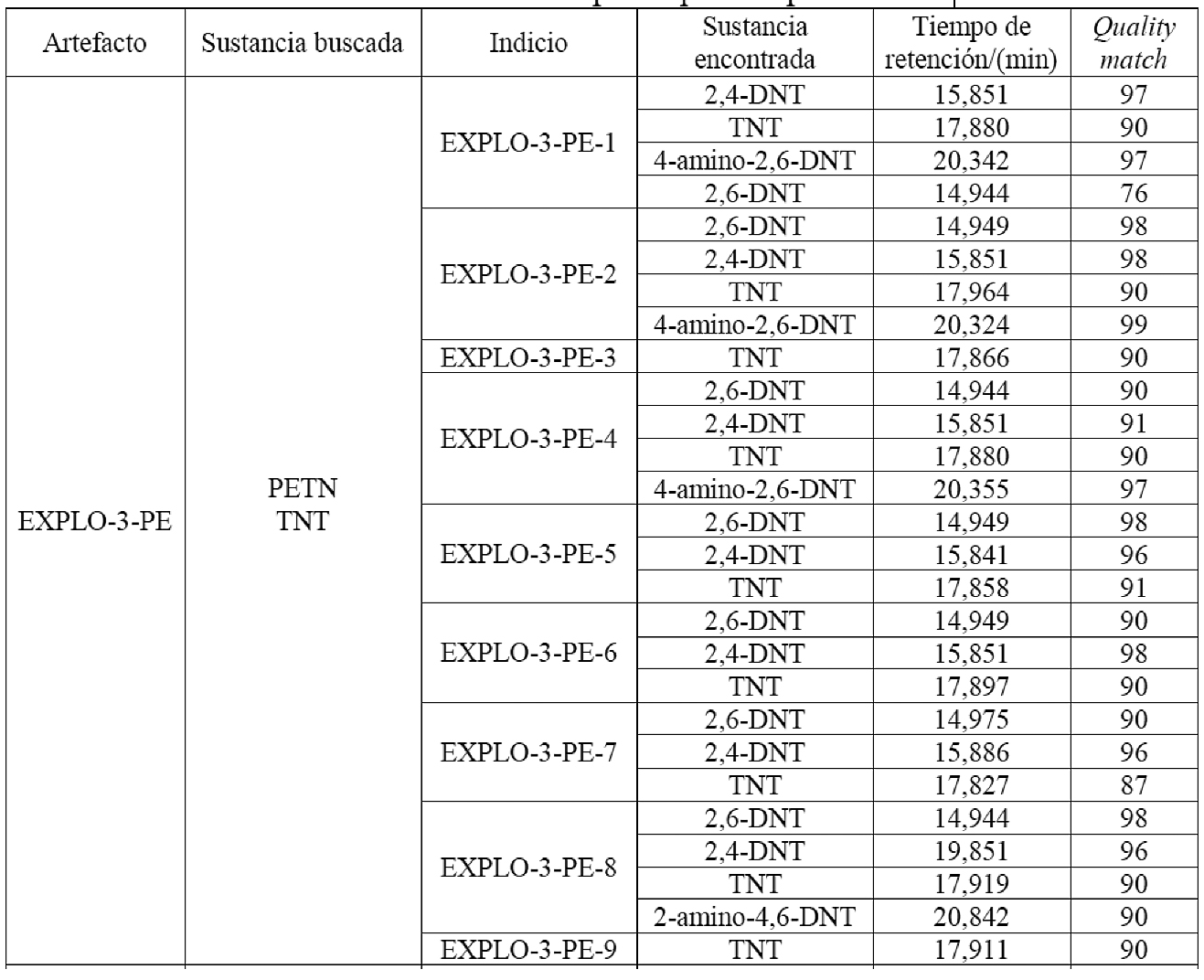
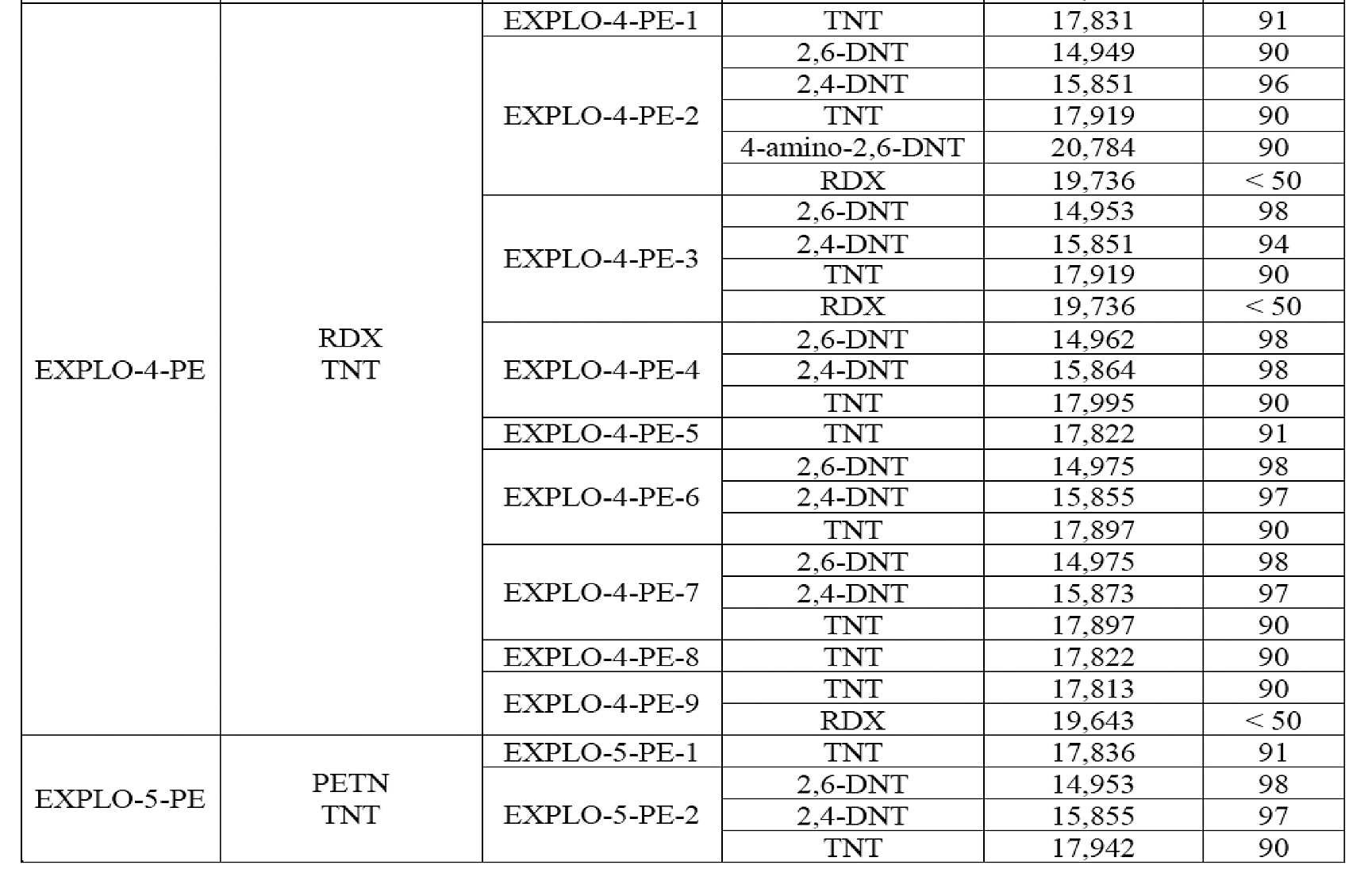
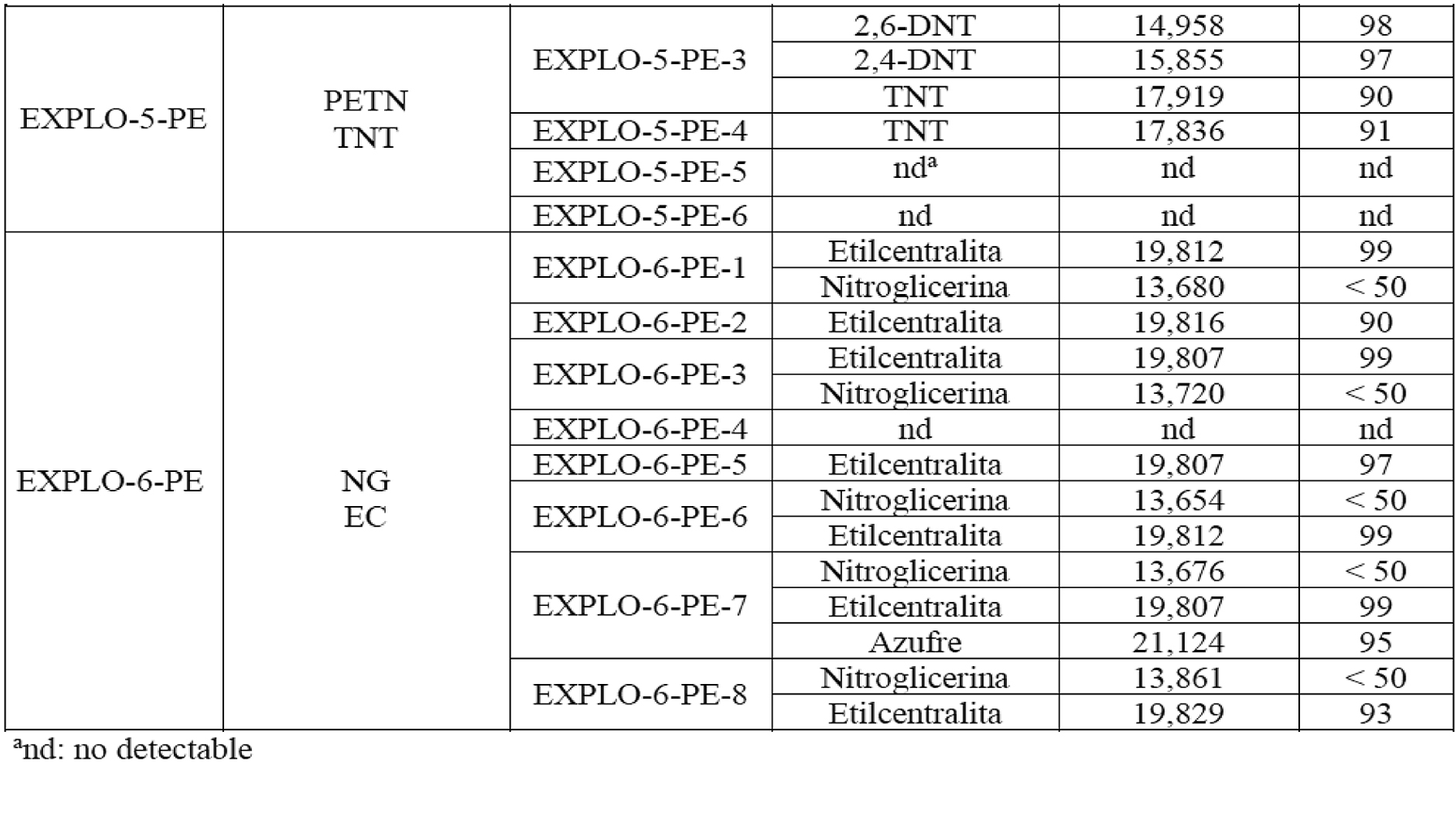
Nota: Los indicios hacen referencia a la cantidad de los fragmentos post-explosión de cada artefacto. Por ejemplo, el artefacto denominado “EXPLO-3-PE” se fragmentó en 8 indicios identificados como EXPLO-3-PE-1, EXPLO-3-PE-2, EXPLO-3-PE-3, EXPLO-3-PE-4, EXPLO-3-PE-5, EXPLO-3-PE-6, EXPLO-3-PE-7 y EXPLO-3-PE-8.
1 Química industrial. Departamento de Ciencias Forenses del Organismo de Investigación Judicial, Costa Rica. Correo electrónico: stephy1608@gmail.com.
2 Químico puro. Departamento de Ciencias Forenses del Organismo de Investigación Judicial. Costa Rica.
3 Químico puro. Escuela de Química, Universidad Nacional, Costa Rica.
Correo electrónico: efrainsolism@gmail.com.
4 Químico puro. Escuela de Química, Universidad Nacional, Costa Rica.
Correo electrónico: gilberto.piedra.marin@una.cr  https://orcid.org/0000-0003-1332-236X
https://orcid.org/0000-0003-1332-236X
5 Químico industrial. Escuela de Química, Universidad Nacional, Costa Rica.
Correo electrónico: jose.mora.barrantes@una.cr https://orcid.org/0000-0002-0409-5276
6 Químico industrial. Escuela de Química, Universidad Nacional, Costa Rica.
Correo electrónico: jose.sibaja.brenes@una.cr https://orcid.org/0000-0002-7056-2717
7 Químico industrial. Escuela de Química, Universidad Nacional, Costa Rica. Correo electrónico: henry.borbon.alpizar@una.cr https://orcid.org/0000-0003-0869-4126